2024
|
Barbieri F; Carlen V; Martina MG; Sannio F; Cancade S; Perini C; Restori M; Crespan E; Maga G; Docquier JD; Cagno V; Radi M. 4-Trifluoromethyl bithiazoles as broad-spectrum antimicrobial agents for virus-related bacterial infections or co-infections Journal Article In: RSC medicinal chemistry, vol. 15, iss. 5, pp. 1589-1600, 2024. @article{%a1.%Y_159,
title = {4-Trifluoromethyl bithiazoles as broad-spectrum antimicrobial agents for virus-related bacterial infections or co-infections},
author = {Barbieri F and Carlen V and Martina MG and Sannio F and Cancade S and Perini C and Restori M and Crespan E and Maga G and Docquier JD and Cagno V and Radi M.},
url = {https://pubs.rsc.org/en/content/articlelanding/2024/md/d3md00686g},
doi = {10.1039/d3md00686g},
year = {2024},
date = {2024-05-28},
journal = {RSC medicinal chemistry},
volume = {15},
issue = {5},
pages = {1589-1600},
abstract = {Respiratory tract infections involving a variety of microorganisms such as viruses, bacteria, and fungi are a prominent cause of morbidity and mortality globally, exacerbating various pre-existing respiratory and non-respiratory conditions. Moreover, the ability of bacteria and viruses to coexist might impact the development and severity of lung infections, promoting bacterial colonization and subsequent disease exacerbation. Secondary bacterial infections following viral infections represent a complex challenge to be overcome from a therapeutic point of view. We report herein our efforts in the development of new bithiazole derivatives showing broad-spectrum antimicrobial activity against both viruses and bacteria. A series of 4-trifluoromethyl bithiazole analogues was synthesized and screened against selected viruses (hRVA16, EVD68, and ZIKV) and a panel of Gram-positive and Gram-negative bacteria. Among them, two promising broad-spectrum antimicrobial compounds (8a and 8j) have been identified: both compounds showed low micromolar activity against all tested viruses, 8a showed synergistic activity against E. coli and A. baumannii in the presence of a subinhibitory concentration of colistin, while 8j showed a broader spectrum of activity against Gram-positive and Gram-negative bacteria. Activity against antibiotic-resistant clinical isolates is also reported. Given the ever-increasing need to adequately address viral and bacterial infections or co-infections, this study paves the way for the development of new agents with broad antimicrobial properties and synergistic activity with common antivirals and antibacterials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Respiratory tract infections involving a variety of microorganisms such as viruses, bacteria, and fungi are a prominent cause of morbidity and mortality globally, exacerbating various pre-existing respiratory and non-respiratory conditions. Moreover, the ability of bacteria and viruses to coexist might impact the development and severity of lung infections, promoting bacterial colonization and subsequent disease exacerbation. Secondary bacterial infections following viral infections represent a complex challenge to be overcome from a therapeutic point of view. We report herein our efforts in the development of new bithiazole derivatives showing broad-spectrum antimicrobial activity against both viruses and bacteria. A series of 4-trifluoromethyl bithiazole analogues was synthesized and screened against selected viruses (hRVA16, EVD68, and ZIKV) and a panel of Gram-positive and Gram-negative bacteria. Among them, two promising broad-spectrum antimicrobial compounds (8a and 8j) have been identified: both compounds showed low micromolar activity against all tested viruses, 8a showed synergistic activity against E. coli and A. baumannii in the presence of a subinhibitory concentration of colistin, while 8j showed a broader spectrum of activity against Gram-positive and Gram-negative bacteria. Activity against antibiotic-resistant clinical isolates is also reported. Given the ever-increasing need to adequately address viral and bacterial infections or co-infections, this study paves the way for the development of new agents with broad antimicrobial properties and synergistic activity with common antivirals and antibacterials. |
2023
|
Contadini C; Cirotti C; Carbone A; Norouzi M; Cianciusi A; Crespan E; Perini C; Maga G; Barilà D; Musumeci F; Schenone S Identification and Biological Characterization of the Pyrazolo[3,4- d]pyrimidine Derivative SI388 Active as Src Inhibitor Journal Article In: Pharmaceuticals - Basel, vol. 16, iss. 7, pp. 958, 2023. @article{%a1.%Yb_98,
title = {Identification and Biological Characterization of the Pyrazolo[3,4- d]pyrimidine Derivative SI388 Active as Src Inhibitor},
author = {Contadini C and Cirotti C and Carbone A and Norouzi M and Cianciusi A and Crespan E and Perini C and Maga G and Barilà D and Musumeci F and Schenone S},
url = {https://www.mdpi.com/1424-8247/16/7/958},
doi = {10.3390/ph16070958},
year = {2023},
date = {2023-08-08},
journal = {Pharmaceuticals - Basel},
volume = {16},
issue = {7},
pages = {958},
abstract = {Src is a non-receptor tyrosine kinase (TK) whose involvement in cancer, including glioblastoma (GBM), has been extensively demonstrated. In this context, we started from our in-house library of pyrazolo[3,4-d]pyrimidines that are active as Src and/or Bcr-Abl TK inhibitors and performed a lead optimization study to discover a new generation derivative that is suitable for Src kinase targeting. We synthesized a library of 19 compounds, 2a-s. Among these, compound 2a (SI388) was identified as the most potent Src inhibitor. Based on the cell-free results, we investigated the effect of SI388 in 2D and 3D GBM cellular models. Interestingly, SI388 significantly inhibits Src kinase, and therefore affects cell viability, tumorigenicity and enhances cancer cell sensitivity to ionizing radiation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Src is a non-receptor tyrosine kinase (TK) whose involvement in cancer, including glioblastoma (GBM), has been extensively demonstrated. In this context, we started from our in-house library of pyrazolo[3,4-d]pyrimidines that are active as Src and/or Bcr-Abl TK inhibitors and performed a lead optimization study to discover a new generation derivative that is suitable for Src kinase targeting. We synthesized a library of 19 compounds, 2a-s. Among these, compound 2a (SI388) was identified as the most potent Src inhibitor. Based on the cell-free results, we investigated the effect of SI388 in 2D and 3D GBM cellular models. Interestingly, SI388 significantly inhibits Src kinase, and therefore affects cell viability, tumorigenicity and enhances cancer cell sensitivity to ionizing radiation. |
Lodola C; Secchi M; Sinigiani V; De Palma A; Rossi R; Perico D; Mauri PL; Maga G Interaction of SARS-CoV-2 Nucleocapsid Protein and Human RNA Helicases DDX1 and DDX3X Modulates Their Activities on Double-Stranded RNA Journal Article In: International journal of molecular sciences, vol. 24, iss. 6, pp. 5784, 2023. @article{%a1.%Yb__97,
title = {Interaction of SARS-CoV-2 Nucleocapsid Protein and Human RNA Helicases DDX1 and DDX3X Modulates Their Activities on Double-Stranded RNA},
author = {Lodola C and Secchi M and Sinigiani V and De Palma A and Rossi R and Perico D and Mauri PL and Maga G},
url = {https://www.mdpi.com/1422-0067/24/6/5784},
doi = {10.3390/ijms24065784},
year = {2023},
date = {2023-08-08},
journal = {International journal of molecular sciences},
volume = {24},
issue = {6},
pages = {5784},
abstract = {The nucleocapsid protein Np of SARS-CoV-2 is involved in the replication, transcription, and packaging of the viral genome, but it also plays a role in the modulation of the host cell innate immunity and inflammation response. Ectopic expression of Np alone was able to induce significant changes in the proteome of human cells. The cellular RNA helicase DDX1 was among the proteins whose levels were increased by Np expression. DDX1 and its related helicase DDX3X were found to physically interact with Np and to increase 2- to 4-fold its affinity for double-stranded RNA in a helicase-independent manner. Conversely, Np inhibited the RNA helicase activity of both proteins. These functional interactions among Np and DDX1 and DDX3X highlight novel possible roles played by these host RNA helicases in the viral life cycle.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The nucleocapsid protein Np of SARS-CoV-2 is involved in the replication, transcription, and packaging of the viral genome, but it also plays a role in the modulation of the host cell innate immunity and inflammation response. Ectopic expression of Np alone was able to induce significant changes in the proteome of human cells. The cellular RNA helicase DDX1 was among the proteins whose levels were increased by Np expression. DDX1 and its related helicase DDX3X were found to physically interact with Np and to increase 2- to 4-fold its affinity for double-stranded RNA in a helicase-independent manner. Conversely, Np inhibited the RNA helicase activity of both proteins. These functional interactions among Np and DDX1 and DDX3X highlight novel possible roles played by these host RNA helicases in the viral life cycle. |
Poggialini F; Vagaggini C; Brai A; Pasqualini C; Crespan E; Maga G; Perini C; Cabella N; Botta L; Musumeci F; Frosini M; Schenone S; Dreassi E Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4- d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme Journal Article In: Pharmaceutics, vol. 15, iss. 2, pp. 453, 2023. @article{%a1.%Yb_75,
title = {Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4- d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme},
author = {Poggialini F and Vagaggini C and Brai A and Pasqualini C and Crespan E and Maga G and Perini C and Cabella N and Botta L and Musumeci F and Frosini M and Schenone S and Dreassi E},
url = {https://www.mdpi.com/1999-4923/15/2/453},
doi = {10.3390/pharmaceutics15020453},
year = {2023},
date = {2023-03-08},
journal = {Pharmaceutics},
volume = {15},
issue = {2},
pages = {453},
abstract = {The therapeutic use of tyrosine kinase inhibitors (TKIs) represents one of the successful strategies for the treatment of glioblastoma (GBM). Pyrazolo[3,4-d]pyrimidines have already been reported as promising small molecules active as c-Src/Abl dual inhibitors. Herein, we present a series of pyrazolo[3,4-d]pyrimidine derivatives, selected from our in-house library, to identify a promising candidate active against GBM. The inhibitory activity against c-Src and Abl was investigated, and the antiproliferative profile against four GBM cell lines was studied. For the most active compounds endowed with antiproliferative efficacy in the low-micromolar range, the effects toward nontumoral, healthy cell lines (fibroblasts FIBRO 2-93 and keratinocytes HaCaT) was investigated. Lastly, the in silico and in vitro ADME properties of all compounds were also assessed. Among the tested compounds, the promising inhibitory activity against c-Src and Abl (Ki 3.14 µM and 0.44 µM, respectively), the irreversible, apoptotic-mediated death toward U-87, LN18, LN229, and DBTRG GBM cell lines (IC50 6.8 µM, 10.8 µM, 6.9 µM, and 8.5 µM, respectively), the significant reduction in GBM cell migration, the safe profile toward FIBRO 2-93 and HaCaT healthy cell lines (CC50 91.7 µM and 126.5 µM, respectively), the high metabolic stability, and the excellent passive permeability across gastrointestinal and blood-brain barriers led us to select compound 5 for further in vivo assays.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The therapeutic use of tyrosine kinase inhibitors (TKIs) represents one of the successful strategies for the treatment of glioblastoma (GBM). Pyrazolo[3,4-d]pyrimidines have already been reported as promising small molecules active as c-Src/Abl dual inhibitors. Herein, we present a series of pyrazolo[3,4-d]pyrimidine derivatives, selected from our in-house library, to identify a promising candidate active against GBM. The inhibitory activity against c-Src and Abl was investigated, and the antiproliferative profile against four GBM cell lines was studied. For the most active compounds endowed with antiproliferative efficacy in the low-micromolar range, the effects toward nontumoral, healthy cell lines (fibroblasts FIBRO 2-93 and keratinocytes HaCaT) was investigated. Lastly, the in silico and in vitro ADME properties of all compounds were also assessed. Among the tested compounds, the promising inhibitory activity against c-Src and Abl (Ki 3.14 µM and 0.44 µM, respectively), the irreversible, apoptotic-mediated death toward U-87, LN18, LN229, and DBTRG GBM cell lines (IC50 6.8 µM, 10.8 µM, 6.9 µM, and 8.5 µM, respectively), the significant reduction in GBM cell migration, the safe profile toward FIBRO 2-93 and HaCaT healthy cell lines (CC50 91.7 µM and 126.5 µM, respectively), the high metabolic stability, and the excellent passive permeability across gastrointestinal and blood-brain barriers led us to select compound 5 for further in vivo assays. |
2022
|
Princiotto S; Musso L; Manetti F; Marcellini V; Maga G; Crespan E; Perini C; Zaffaroni N; Beretta GL; Dallavalle S Synthesis and biological activity evaluation of 3-(hetero) arylideneindolin-2-ones as potential c-Src inhibitors Journal Article In: Journal of enzyme inhibition and medicinal chemistry, vol. 37, iss. 1, pp. 2382-2394, 2022. @article{%a1.%Yb_41,
title = {Synthesis and biological activity evaluation of 3-(hetero) arylideneindolin-2-ones as potential c-Src inhibitors},
author = {Princiotto S and Musso L and Manetti F and Marcellini V and Maga G and Crespan E and Perini C and Zaffaroni N and Beretta GL and Dallavalle S},
url = {https://www.tandfonline.com/doi/full/10.1080/14756366.2022.2117317},
doi = {10.1080/14756366.2022.2117317},
year = {2022},
date = {2022-09-05},
journal = {Journal of enzyme inhibition and medicinal chemistry},
volume = {37},
issue = {1},
pages = {2382-2394},
abstract = {Inhibition of c-Src is considered one of the most studied approaches to cancer treatment, with several heterocyclic compounds approved during the last 15 years as chemotherapeutic agents. Starting from the biological evaluation of an in-house collection of small molecules, indolinone was selected as the most promising scaffold. In this work, several functionalised indolinones were synthesised and their inhibitory potency and cytotoxic activity were assayed. The pharmacological profile of the most active compounds, supported by molecular modelling studies, revealed that the presence of an amino group increased the affinity towards the ATP-binding site of c-Src. At the same time, bulkier derivatizations seemed to improve the interactions within the enzymatic pocket. Overall, these data represent an early stage towards the optimisation of new, easy-to-be functionalised indolinones as potential c-Src inhibitors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Inhibition of c-Src is considered one of the most studied approaches to cancer treatment, with several heterocyclic compounds approved during the last 15 years as chemotherapeutic agents. Starting from the biological evaluation of an in-house collection of small molecules, indolinone was selected as the most promising scaffold. In this work, several functionalised indolinones were synthesised and their inhibitory potency and cytotoxic activity were assayed. The pharmacological profile of the most active compounds, supported by molecular modelling studies, revealed that the presence of an amino group increased the affinity towards the ATP-binding site of c-Src. At the same time, bulkier derivatizations seemed to improve the interactions within the enzymatic pocket. Overall, these data represent an early stage towards the optimisation of new, easy-to-be functionalised indolinones as potential c-Src inhibitors. |
Secchi M; Lodola C; Garbelli A; Bione S; Maga G DEAD-Box RNA Helicases DDX3X and DDX5 as Oncogenes or Oncosuppressors: A Network Perspective Journal Article In: Cancers (Basel), vol. 14, iss. 15, pp. 3820, 2022. @article{%a1.%Yb_37,
title = {DEAD-Box RNA Helicases DDX3X and DDX5 as Oncogenes or Oncosuppressors: A Network Perspective},
author = {Secchi M and Lodola C and Garbelli A and Bione S and Maga G},
url = {https://www.mdpi.com/2072-6694/14/15/3820},
doi = {10.3390/cancers14153820},
year = {2022},
date = {2022-08-18},
journal = {Cancers (Basel)},
volume = {14},
issue = {15},
pages = {3820},
abstract = {RNA helicases of the DEAD-box family are involved in several metabolic pathways, from transcription and translation to cell proliferation, innate immunity and stress response. Given their multiple roles, it is not surprising that their deregulation or mutation is linked to different pathological conditions, including cancer. However, while in some cases the loss of function of a given DEAD-box helicase promotes tumor transformation, indicating an oncosuppressive role, in other contexts the overexpression of the same enzyme favors cancer progression, thus acting as a typical oncogene. The roles of two well-characterized members of this family, DDX3X and DDX5, as both oncogenes and oncosuppressors have been documented in several cancer types. Understanding the interplay of the different cellular contexts, as defined by the molecular interaction networks of DDX3X and DDX5 in different tumors, with the cancer-specific roles played by these proteins could help to explain their apparently conflicting roles as cancer drivers or suppressors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
RNA helicases of the DEAD-box family are involved in several metabolic pathways, from transcription and translation to cell proliferation, innate immunity and stress response. Given their multiple roles, it is not surprising that their deregulation or mutation is linked to different pathological conditions, including cancer. However, while in some cases the loss of function of a given DEAD-box helicase promotes tumor transformation, indicating an oncosuppressive role, in other contexts the overexpression of the same enzyme favors cancer progression, thus acting as a typical oncogene. The roles of two well-characterized members of this family, DDX3X and DDX5, as both oncogenes and oncosuppressors have been documented in several cancer types. Understanding the interplay of the different cellular contexts, as defined by the molecular interaction networks of DDX3X and DDX5 in different tumors, with the cancer-specific roles played by these proteins could help to explain their apparently conflicting roles as cancer drivers or suppressors. |
Di Maria S; Picarazzi F; Mori M; Cianciusi A; Carbone A; Crespan E; Perini C; Sabetta S; Deplano S; Poggialini F; Molinari A; Aronne R; Maccioni E; Maga G; Angelucci A; Schenone S; Musumeci F; Dreassi E Novel pyrazolo[3,4-d]pyrimidines as dual Src/Bcr-Abl kinase inhibitors: Synthesis and biological evaluation for chronic myeloid leukemia treatment. Journal Article In: Bioorganic chemistry, vol. 128, pp. 106071, 2022. @article{%a1.%Yb_50,
title = {Novel pyrazolo[3,4-d]pyrimidines as dual Src/Bcr-Abl kinase inhibitors: Synthesis and biological evaluation for chronic myeloid leukemia treatment. },
author = {Di Maria S and Picarazzi F and Mori M and Cianciusi A and Carbone A and Crespan E and Perini C and Sabetta S and Deplano S and Poggialini F and Molinari A and Aronne R and Maccioni E and Maga G and Angelucci A and Schenone S and Musumeci F and Dreassi E},
url = {https://www.sciencedirect.com/science/article/pii/S0045206822004771?via%3Dihub},
doi = {10.1016/j.bioorg.2022.106071},
year = {2022},
date = {2022-03-30},
journal = {Bioorganic chemistry},
volume = {128},
pages = {106071},
abstract = {The Bcr-Abl tyrosine kinase (TK) is the molecular hallmark of chronic myeloid leukemia (CML). Src is another TK kinase whose involvement in CML was widely demonstrated. Small molecules active as dual Src/Bcr-Abl inhibitors emerged as effective targeted therapies for CML and a few compounds are currently in clinical use. In this study, we applied a target-oriented approach to identify a family of pyrazolo[3,4-d]pyrimidines as dual Src/Bcr-Abl inhibitors as anti-leukemia agents. Considering the high homology between Src and Bcr-Abl, in-house Src inhibitors 8a-l and new analogue compounds 9a-n were screened as dual Src/Bcr-Abl inhibitors. The antiproliferative activity on K562 CML cells and the ADME profile were determined for the most promising compounds. Molecular modeling studies elucidated the binding mode of the inhibitors into the Bcr-Abl (wt) catalytic pocket. Compounds 8j and 8k showed nanomolar activities in enzymatic and cellular assays, together with favorable ADME properties, emerging as promising candidates for CML therapy. Finally, derivatives 9j and 9k, emerging as valuable inhibitors of the most aggressive Bcr-Abl mutation, T315I, constitute a good starting point in the search for compounds able to treat drug-resistant forms of CML. Overall, this study allowed us to identify more potent compounds than those previously reported by the group, marking a step forward in searching for new antileukemic agents.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The Bcr-Abl tyrosine kinase (TK) is the molecular hallmark of chronic myeloid leukemia (CML). Src is another TK kinase whose involvement in CML was widely demonstrated. Small molecules active as dual Src/Bcr-Abl inhibitors emerged as effective targeted therapies for CML and a few compounds are currently in clinical use. In this study, we applied a target-oriented approach to identify a family of pyrazolo[3,4-d]pyrimidines as dual Src/Bcr-Abl inhibitors as anti-leukemia agents. Considering the high homology between Src and Bcr-Abl, in-house Src inhibitors 8a-l and new analogue compounds 9a-n were screened as dual Src/Bcr-Abl inhibitors. The antiproliferative activity on K562 CML cells and the ADME profile were determined for the most promising compounds. Molecular modeling studies elucidated the binding mode of the inhibitors into the Bcr-Abl (wt) catalytic pocket. Compounds 8j and 8k showed nanomolar activities in enzymatic and cellular assays, together with favorable ADME properties, emerging as promising candidates for CML therapy. Finally, derivatives 9j and 9k, emerging as valuable inhibitors of the most aggressive Bcr-Abl mutation, T315I, constitute a good starting point in the search for compounds able to treat drug-resistant forms of CML. Overall, this study allowed us to identify more potent compounds than those previously reported by the group, marking a step forward in searching for new antileukemic agents. |
Cesarini S; Vicenti I; Poggialini F; Secchi M; Giammarino F; Varasi I; Lodola C; Zazzi M; Dreassi E; Maga G; Botta L; Saladino R Privileged Scaffold Decoration for the Identification of the First Trisubstituted Triazine with Anti-SARS-CoV-2 Activity Journal Article In: Molecules, vol. 27, iss. 24, pp. 8829, 2022. @article{%a1.%Yb_46,
title = {Privileged Scaffold Decoration for the Identification of the First Trisubstituted Triazine with Anti-SARS-CoV-2 Activity},
author = {Cesarini S and Vicenti I and Poggialini F and Secchi M and Giammarino F and Varasi I and Lodola C and Zazzi M and Dreassi E and Maga G and Botta L and Saladino R},
url = {https://www.mdpi.com/1420-3049/27/24/8829},
doi = {10.3390/molecules27248829},
year = {2022},
date = {2022-03-25},
journal = {Molecules},
volume = {27},
issue = {24},
pages = {8829},
abstract = {Current therapy against severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) are based on the use of Remdesivir 1, Molnupiravir 2, and the recently identified Nirmatrelvir 3. Unfortunately, these three drugs showed some limitations regarding potency and possible drug-drug interactions. A series of derivatives coming from a decoration approach of the privileged scaffold s-triazines were synthesized and evaluated against SAR-CoV-2. One derivative emerged as the hit of the series for its micromolar antiviral activity and low cytotoxicity. Mode of action and pharmacokinetic in vitro preliminary studies further confirm the role as candidates for a future optimization campaign of the most active derivative identified with this work.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Current therapy against severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) are based on the use of Remdesivir 1, Molnupiravir 2, and the recently identified Nirmatrelvir 3. Unfortunately, these three drugs showed some limitations regarding potency and possible drug-drug interactions. A series of derivatives coming from a decoration approach of the privileged scaffold s-triazines were synthesized and evaluated against SAR-CoV-2. One derivative emerged as the hit of the series for its micromolar antiviral activity and low cytotoxicity. Mode of action and pharmacokinetic in vitro preliminary studies further confirm the role as candidates for a future optimization campaign of the most active derivative identified with this work. |
2021
|
Brai A; Riva V; Clementi L; Falsitta L; Zamperini C; Sinigiani V; Festuccia C; Sabetta S; Aiello D; Roselli C; Garbelli A; Trivisani CI; Maccari L; Bugli F; Sanguinetti M; Calandro P; Chiariello M; Quaranta P; Botta L; Angelucci A; Maga G; Botta M Targeting DDX3X Helicase Activity with BA103 Shows Promising Therapeutic Effects in Preclinical Glioblastoma Models Journal Article In: Cancers (Basel), vol. 13, no 21, pp. 5569, 2021. @article{%a1:%Yb_60,
title = {Targeting DDX3X Helicase Activity with BA103 Shows Promising Therapeutic Effects in Preclinical Glioblastoma Models},
author = {Brai A and Riva V and Clementi L and Falsitta L and Zamperini C and Sinigiani V and Festuccia C and Sabetta S and Aiello D and Roselli C and Garbelli A and Trivisani CI and Maccari L and Bugli F and Sanguinetti M and Calandro P and Chiariello M and Quaranta P and Botta L and Angelucci A and Maga G and Botta M},
url = {https://www.mdpi.com/2072-6694/13/21/5569},
doi = {10.3390/cancers13215569},
year = {2021},
date = {2021-12-06},
journal = {Cancers (Basel)},
volume = {13},
number = {21},
pages = {5569},
abstract = {DDX3X is an ATP-dependent RNA helicase that has recently attracted interest for its involvement in viral replication and oncogenic progression. Starting from hit compounds previously identified by our group, we have designed and synthesized a new series of DDX3X inhibitors that effectively blocked its helicase activity. These new compounds were able to inhibit the proliferation of cell lines from different cancer types, also in DDX3X low-expressing cancer cell lines. According to the absorption, distribution, metabolism, elimination properties, and antitumoral activity, compound BA103 was chosen to be further investigated in glioblastoma models. BA103 determined a significant reduction in the proliferation and migration of U87 and U251 cells, downregulating the oncogenic protein β-catenin. An in vivo evaluation demonstrated that BA103 was able to reach the brain and reduce the tumor growth in xenograft and orthotopic models without evident side effects. This study represents the first demonstration that DDX3X-targeted small molecules are feasible and promising drugs also in glioblastoma.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
DDX3X is an ATP-dependent RNA helicase that has recently attracted interest for its involvement in viral replication and oncogenic progression. Starting from hit compounds previously identified by our group, we have designed and synthesized a new series of DDX3X inhibitors that effectively blocked its helicase activity. These new compounds were able to inhibit the proliferation of cell lines from different cancer types, also in DDX3X low-expressing cancer cell lines. According to the absorption, distribution, metabolism, elimination properties, and antitumoral activity, compound BA103 was chosen to be further investigated in glioblastoma models. BA103 determined a significant reduction in the proliferation and migration of U87 and U251 cells, downregulating the oncogenic protein β-catenin. An in vivo evaluation demonstrated that BA103 was able to reach the brain and reduce the tumor growth in xenograft and orthotopic models without evident side effects. This study represents the first demonstration that DDX3X-targeted small molecules are feasible and promising drugs also in glioblastoma. |
Grazia Martina M; Vicenti I; Bauer L; Crespan E; Rango E; Boccuto A; Olivieri N; Incerti M; Zwaagstra M; Allodi M; Bertoni S; Dreassi E; Zazzi M; van Kuppeveld FJM; Maga G; Radi M Bithiazole Inhibitors of Phosphatidylinositol 4-Kinase (PI4KIIIBETA) as Broad-Spectrum Antivirals Blocking the Replication of SARS-CoV-2, Zika Virus, and Human Rhinoviruses Journal Article In: ChemMedChem, vol. 16, iss. 23, no 3548, pp. 3552, 2021. @article{%a1:%Ybvwb,
title = {Bithiazole Inhibitors of Phosphatidylinositol 4-Kinase (PI4KIIIBETA) as Broad-Spectrum Antivirals Blocking the Replication of SARS-CoV-2, Zika Virus, and Human Rhinoviruses},
author = {{Grazia Martina M} and Vicenti I and Bauer L and Crespan E and Rango E and Boccuto A and Olivieri N and Incerti M and Zwaagstra M and Allodi M and Bertoni S and Dreassi E and Zazzi M and van Kuppeveld FJM and Maga G and Radi M},
url = {https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.202100483},
doi = {10.1002/cmdc.202100483},
year = {2021},
date = {2021-11-08},
urldate = {2021-11-08},
journal = {ChemMedChem},
volume = {16},
number = {3548},
issue = {23},
pages = {3552},
abstract = {Over half a century since the description of the first antiviral drug, "old" re-emerging viruses and "new" emerging viruses still represent a serious threat to global health. Their high mutation rate and rapid selection of resistance toward common antiviral drugs, together with the increasing number of co-infections, make the war against viruses quite challenging. Herein we report a host-targeted approach, based on the inhibition of the lipid kinase PI4KIIIβ, as a promising strategy for inhibiting the replication of multiple viruses hijacking this protein. We show that bithiazole inhibitors of PI4KIIIβ block the replication of human rhinoviruses (hRV), Zika virus (ZIKV) and SARS-CoV-2 at low micromolar and sub-micromolar concentrations. However, while the anti-hRV/ZIKV activity can be directly linked to PI4KIIIβ inhibition, the role of PI4KIIIβ in SARS-CoV-2 entry/replication is debated.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Over half a century since the description of the first antiviral drug, "old" re-emerging viruses and "new" emerging viruses still represent a serious threat to global health. Their high mutation rate and rapid selection of resistance toward common antiviral drugs, together with the increasing number of co-infections, make the war against viruses quite challenging. Herein we report a host-targeted approach, based on the inhibition of the lipid kinase PI4KIIIβ, as a promising strategy for inhibiting the replication of multiple viruses hijacking this protein. We show that bithiazole inhibitors of PI4KIIIβ block the replication of human rhinoviruses (hRV), Zika virus (ZIKV) and SARS-CoV-2 at low micromolar and sub-micromolar concentrations. However, while the anti-hRV/ZIKV activity can be directly linked to PI4KIIIβ inhibition, the role of PI4KIIIβ in SARS-CoV-2 entry/replication is debated. |
Vicenti I; Martina MG; Boccuto A; De Angelis M; Giavarini G; Dragoni F; Marchi S; Trombetta CM; Crespan E; Maga G; Eydoux C; Decroly E; Montomoli E; Nencioni L; Zazzi M; Radi M System-oriented optimization of multi-target 2,6-diaminopurine derivatives: Easily accessible broad-spectrum antivirals active against flaviviruses, influenza virus and SARS-CoV-2. Journal Article In: European journal of medicinal chemistry, vol. 224, pp. 113683, 2021. @article{%a1:%Ybv,
title = {System-oriented optimization of multi-target 2,6-diaminopurine derivatives: Easily accessible broad-spectrum antivirals active against flaviviruses, influenza virus and SARS-CoV-2. },
author = {Vicenti I and Martina MG and Boccuto A and De Angelis M and Giavarini G and Dragoni F and Marchi S and Trombetta CM and Crespan E and Maga G and Eydoux C and Decroly E and Montomoli E and Nencioni L and Zazzi M and Radi M},
url = {https://www.sciencedirect.com/science/article/pii/S0223523421005328?via%3Dihub},
doi = {10.1016/j.ejmech.2021.113683},
year = {2021},
date = {2021-08-25},
journal = {European journal of medicinal chemistry},
volume = {224},
pages = {113683},
abstract = {he worldwide circulation of different viruses coupled with the increased frequency and diversity of new outbreaks, strongly highlight the need for new antiviral drugs to quickly react against potential pandemic pathogens. Broad-spectrum antiviral agents (BSAAs) represent the ideal option for a prompt response against multiple viruses, new and re-emerging. Starting from previously identified anti-flavivirus hits, we report herein the identification of promising BSAAs by submitting the multi-target 2,6-diaminopurine chemotype to a system-oriented optimization based on phenotypic screening on cell cultures infected with different viruses. Among the synthesized compounds, 6i showed low micromolar potency against Dengue, Zika, West Nile and Influenza A viruses (IC50 = 0.5-5.3 μM) with high selectivity index. Interestingly, 6i also inhibited SARS-CoV-2 replication in different cell lines, with higher potency on Calu-3 cells that better mimic the SARS-CoV-2 infection in vivo (IC50 = 0.5 μM, SI = 240). The multi-target effect of 6i on flavivirus replication was also analyzed in whole cell studies (in vitro selection and immunofluorescence) and against isolated host/viral targets.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
he worldwide circulation of different viruses coupled with the increased frequency and diversity of new outbreaks, strongly highlight the need for new antiviral drugs to quickly react against potential pandemic pathogens. Broad-spectrum antiviral agents (BSAAs) represent the ideal option for a prompt response against multiple viruses, new and re-emerging. Starting from previously identified anti-flavivirus hits, we report herein the identification of promising BSAAs by submitting the multi-target 2,6-diaminopurine chemotype to a system-oriented optimization based on phenotypic screening on cell cultures infected with different viruses. Among the synthesized compounds, 6i showed low micromolar potency against Dengue, Zika, West Nile and Influenza A viruses (IC50 = 0.5-5.3 μM) with high selectivity index. Interestingly, 6i also inhibited SARS-CoV-2 replication in different cell lines, with higher potency on Calu-3 cells that better mimic the SARS-CoV-2 infection in vivo (IC50 = 0.5 μM, SI = 240). The multi-target effect of 6i on flavivirus replication was also analyzed in whole cell studies (in vitro selection and immunofluorescence) and against isolated host/viral targets. |
Dede M; Napolitano S; Melati A; Pirota V; Maga G; Crespan E High Flexibility of RNaseH2 Catalytic Activity with Respect to Non-Canonical DNA Structures Journal Article In: International journal of molecular sciences, vol. 22, no 10, pp. 5201, 2021. @article{%a1:%Yb,
title = {High Flexibility of RNaseH2 Catalytic Activity with Respect to Non-Canonical DNA Structures},
author = {Dede M and Napolitano S and Melati A and Pirota V and Maga G and Crespan E},
url = {https://www.mdpi.com/1422-0067/22/10/5201},
doi = {10.3390/ijms22105201},
year = {2021},
date = {2021-06-08},
journal = {International journal of molecular sciences},
volume = {22},
number = {10},
pages = {5201},
abstract = {Ribonucleotides misincorporated in the human genome are the most abundant DNA lesions. The 2'-hydroxyl group makes them prone to spontaneous hydrolysis, potentially resulting in strand breaks. Moreover, their presence may decrease the rate of DNA replication causing replicative fork stalling and collapse. Ribonucleotide removal is initiated by Ribonuclease H2 (RNase H2), the key player in Ribonucleotide Excision Repair (RER). Its absence leads to embryonic lethality in mice, while mutations decreasing its activity cause Aicardi-Goutières syndrome. DNA geometry can be altered by DNA lesions or by peculiar sequences forming secondary structures, like G-quadruplex (G4) and trinucleotide repeats (TNR) hairpins, which significantly differ from canonical B-form. Ribonucleotides pairing to lesioned nucleotides, or incorporated within non-B DNA structures could avoid RNase H2 recognition, potentially contributing to genome instability. In this work, we investigate the ability of RNase H2 to process misincorporated ribonucleotides in a panel of DNA substrates showing different geometrical features. RNase H2 proved to be a flexible enzyme, recognizing as a substrate the majority of the constructs we generated. However, some geometrical features and non-canonical DNA structures severely impaired its activity, suggesting a relevant role of misincorporated ribonucleotides in the physiological instability of specific DNA sequences.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ribonucleotides misincorporated in the human genome are the most abundant DNA lesions. The 2'-hydroxyl group makes them prone to spontaneous hydrolysis, potentially resulting in strand breaks. Moreover, their presence may decrease the rate of DNA replication causing replicative fork stalling and collapse. Ribonucleotide removal is initiated by Ribonuclease H2 (RNase H2), the key player in Ribonucleotide Excision Repair (RER). Its absence leads to embryonic lethality in mice, while mutations decreasing its activity cause Aicardi-Goutières syndrome. DNA geometry can be altered by DNA lesions or by peculiar sequences forming secondary structures, like G-quadruplex (G4) and trinucleotide repeats (TNR) hairpins, which significantly differ from canonical B-form. Ribonucleotides pairing to lesioned nucleotides, or incorporated within non-B DNA structures could avoid RNase H2 recognition, potentially contributing to genome instability. In this work, we investigate the ability of RNase H2 to process misincorporated ribonucleotides in a panel of DNA substrates showing different geometrical features. RNase H2 proved to be a flexible enzyme, recognizing as a substrate the majority of the constructs we generated. However, some geometrical features and non-canonical DNA structures severely impaired its activity, suggesting a relevant role of misincorporated ribonucleotides in the physiological instability of specific DNA sequences. |
Mentegari E; Bertoletti F; Kissova M; Zucca E; Galli S; Tagliavini G; Garbelli A; Maffia A; Bione S; Ferrari E; d'Adda di Fagagna F; Francia S; Sabbioneda S; Chen LY; Lingner J; Bergoglio V; Hoffmann JS; Hubscher U; Crespan E; Maga G A Role for Human DNA Polymerase lambda in Alternative Lengthening of Telomeres Journal Article In: International journal of molecular sciences, vol. 22, no 5, pp. 2365, 2021. @article{%a1:%Y_131,
title = {A Role for Human DNA Polymerase lambda in Alternative Lengthening of Telomeres},
author = {Mentegari E and Bertoletti F and Kissova M and Zucca E and Galli S and Tagliavini G and Garbelli A and Maffia A and Bione S and Ferrari E and {d'Adda di Fagagna F} and Francia S and Sabbioneda S and Chen LY and Lingner J and Bergoglio V and Hoffmann JS and Hubscher U and Crespan E and Maga G},
url = {https://www.mdpi.com/1422-0067/22/5/2365},
doi = {10.3390/ijms22052365},
year = {2021},
date = {2021-03-09},
journal = {International journal of molecular sciences},
volume = {22},
number = {5},
pages = {2365},
abstract = {Telomerase negative cancer cell types use the Alternative Lengthening of Telomeres (ALT) pathway to elongate telomeres ends. Here, we show that silencing human DNA polymerase (Pol lambda) in ALT cells represses ALT activity and induces telomeric stress. In addition, replication stress in the absence of Pol lambda, strongly affects the survival of ALT cells. In vitro, Pol lambda can promote annealing of even a single G-rich telomeric repeat to its complementary strand and use it to prime DNA synthesis. The noncoding telomeric repeat containing RNA TERRA and replication protein A negatively regulate this activity, while the Protection of Telomeres protein 1 (POT1)/TPP1 heterodimer stimulates Pol lambda. Pol lambda associates with telomeres and colocalizes with TPP1 in cells. In summary, our data suggest a role of Pol lambda in the maintenance of telomeres by the ALT mechanism.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Telomerase negative cancer cell types use the Alternative Lengthening of Telomeres (ALT) pathway to elongate telomeres ends. Here, we show that silencing human DNA polymerase (Pol lambda) in ALT cells represses ALT activity and induces telomeric stress. In addition, replication stress in the absence of Pol lambda, strongly affects the survival of ALT cells. In vitro, Pol lambda can promote annealing of even a single G-rich telomeric repeat to its complementary strand and use it to prime DNA synthesis. The noncoding telomeric repeat containing RNA TERRA and replication protein A negatively regulate this activity, while the Protection of Telomeres protein 1 (POT1)/TPP1 heterodimer stimulates Pol lambda. Pol lambda associates with telomeres and colocalizes with TPP1 in cells. In summary, our data suggest a role of Pol lambda in the maintenance of telomeres by the ALT mechanism. |
2020
|
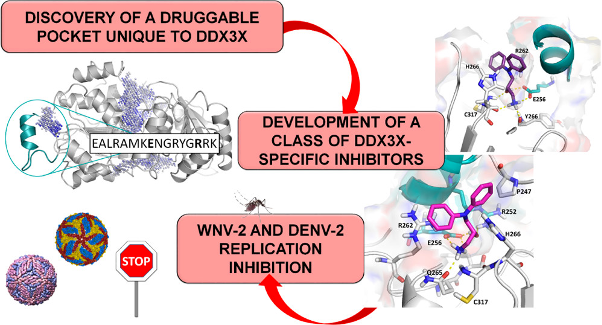
Riva V; Garbelli A; Brai A; Casiraghi F; Fazi R; Trivisani CI; Boccuto A; Saladini F; Vicenti I; Martelli F; Zazzi M; Giannecchini S; Dreassi E; Botta M; Maga G Unique Domain for a Unique Target: Selective Inhibitors of Host Cell DDX3X to Fight Emerging Viruses Journal Article In: Journal of medicinal chemistry, vol. 63, no 17, pp. 9876-9877, 2020. @article{%a1:%Y_5555,
title = {Unique Domain for a Unique Target: Selective Inhibitors of Host Cell DDX3X to Fight Emerging Viruses},
author = {Riva V and Garbelli A and Brai A and Casiraghi F and Fazi R and Trivisani CI and Boccuto A and Saladini F and Vicenti I and Martelli F and Zazzi M and Giannecchini S and Dreassi E and Botta M and Maga G},
url = {https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01039},
doi = {10.1021/acs.jmedchem.0c01039},
year = {2020},
date = {2020-12-17},
urldate = {2020-12-17},
journal = {Journal of medicinal chemistry},
volume = {63},
number = {17},
pages = {9876-9877},
abstract = {• Emerging viruses like dengue, West Nile, chikungunya, and Zika can cause widespread viral epidemics. Developing novel drugs or vaccines against specific targets for each virus is a difficult task. As obligate parasites, all viruses exploit common cellular pathways, providing the possibility to develop broad-spectrum antiviral agents targeting host factors. The human DEAD-box RNA helicase DDX3X is an essential cofactor for viral replication but dispensable for cell viability. Herein, we exploited the presence of a unique structural motif of DDX3X not shared by other cellular enzymes to develop a theoretical model to aid in the design of a novel class of highly selective inhibitors acting against such specific targets, thus limiting off-targeting effects. High-throughput virtual screening led us to identify hit compound 5, endowed with promising antienzymatic activity. To improve its aqueous solubility, 5 and its two enantiomers were synthesized and converted into their corresponding acetate salts (compounds 11, 12, and 13). In vitro mutagenesis and biochemical and cellular assays further confirmed that the developed molecules were selective for DDX3X and were able to suppress replication of West Nile and dengue viruses in infected cells in the micromolar range while showing no toxicity for uninfected cells. These results provide proof of principle for a novel strategy in developing highly selective and broad-spectrum antiviral molecules active against emerging and dangerous viral pathogens. This study paves the way for the development of larger focused libraries targeting such domain to expand SAR studies and fully characterize their mode of interaction.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
• Emerging viruses like dengue, West Nile, chikungunya, and Zika can cause widespread viral epidemics. Developing novel drugs or vaccines against specific targets for each virus is a difficult task. As obligate parasites, all viruses exploit common cellular pathways, providing the possibility to develop broad-spectrum antiviral agents targeting host factors. The human DEAD-box RNA helicase DDX3X is an essential cofactor for viral replication but dispensable for cell viability. Herein, we exploited the presence of a unique structural motif of DDX3X not shared by other cellular enzymes to develop a theoretical model to aid in the design of a novel class of highly selective inhibitors acting against such specific targets, thus limiting off-targeting effects. High-throughput virtual screening led us to identify hit compound 5, endowed with promising antienzymatic activity. To improve its aqueous solubility, 5 and its two enantiomers were synthesized and converted into their corresponding acetate salts (compounds 11, 12, and 13). In vitro mutagenesis and biochemical and cellular assays further confirmed that the developed molecules were selective for DDX3X and were able to suppress replication of West Nile and dengue viruses in infected cells in the micromolar range while showing no toxicity for uninfected cells. These results provide proof of principle for a novel strategy in developing highly selective and broad-spectrum antiviral molecules active against emerging and dangerous viral pathogens. This study paves the way for the development of larger focused libraries targeting such domain to expand SAR studies and fully characterize their mode of interaction. |
Lico C; Santi L; Baschieri S; Noris E; Marusic C; Donini M; Pedrazzini E; Maga G; Franconi R; Di Bonito P; Avesani Plant Molecular Farming as a Strategy Against COVID-19 - The Italian Perspective. Journal Article In: Frontiers in plant science, vol. 11, pp. 609910, 2020. @article{%a1:%Y__486,
title = {Plant Molecular Farming as a Strategy Against COVID-19 - The Italian Perspective. },
author = {Lico C and Santi L and Baschieri S and Noris E and Marusic C and Donini M and Pedrazzini E and Maga G and Franconi R and Di Bonito P and Avesani },
url = {https://www.frontiersin.org/articles/10.3389/fpls.2020.609910/full},
doi = {10.3389/fpls.2020.609910},
year = {2020},
date = {2020-12-01},
journal = {Frontiers in plant science},
volume = {11},
pages = {609910},
abstract = {Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has killed more than 37,000 people in Italy and has caused widespread socioeconomic disruption. Urgent measures are needed to contain and control the virus, particularly diagnostic kits for detection and surveillance, therapeutics to reduce mortality among the severely affected, and vaccines to protect the remaining population. Here we discuss the potential role of plant molecular farming in the rapid and scalable supply of protein antigens as reagents and vaccine candidates, antibodies for virus detection and passive immunotherapy, other therapeutic proteins, and virus-like particles as novel vaccine platforms. We calculate the amount of infrastructure and production capacity needed to deal with predictable subsequent waves of COVID-19 in Italy by pooling expertise in plant molecular farming, epidemiology and the Italian health system. We calculate the investment required in molecular farming infrastructure that would enable us to capitalize on this technology, and provide a roadmap for the development of diagnostic reagents and biopharmaceuticals using molecular farming in plants to complement production methods based on the cultivation of microbes and mammalian cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has killed more than 37,000 people in Italy and has caused widespread socioeconomic disruption. Urgent measures are needed to contain and control the virus, particularly diagnostic kits for detection and surveillance, therapeutics to reduce mortality among the severely affected, and vaccines to protect the remaining population. Here we discuss the potential role of plant molecular farming in the rapid and scalable supply of protein antigens as reagents and vaccine candidates, antibodies for virus detection and passive immunotherapy, other therapeutic proteins, and virus-like particles as novel vaccine platforms. We calculate the amount of infrastructure and production capacity needed to deal with predictable subsequent waves of COVID-19 in Italy by pooling expertise in plant molecular farming, epidemiology and the Italian health system. We calculate the investment required in molecular farming infrastructure that would enable us to capitalize on this technology, and provide a roadmap for the development of diagnostic reagents and biopharmaceuticals using molecular farming in plants to complement production methods based on the cultivation of microbes and mammalian cells. |
Squeglia F; Romano M; Ruggiero A; Maga G; Berisio R Host DDX Helicases as Possible SARS-CoV-2 Proviral Factors: A Structural Overview of Their Hijacking Through Multiple Viral Proteins. Journal Article In: Frontiers in chemistry, vol. 8, pp. 602162, 2020. @article{%a1:%Y__485,
title = {Host DDX Helicases as Possible SARS-CoV-2 Proviral Factors: A Structural Overview of Their Hijacking Through Multiple Viral Proteins.},
author = {Squeglia F and Romano M and Ruggiero A and Maga G and Berisio R},
url = {https://www.frontiersin.org/articles/10.3389/fchem.2020.602162/full#h7},
doi = {10.3389/fchem.2020.602162},
year = {2020},
date = {2020-12-01},
journal = {Frontiers in chemistry},
volume = {8},
pages = {602162},
abstract = {As intracellular parasites, viruses hijack the host cell metabolic machinery for their replication. Among other cellular proteins, the DEAD-box (DDX) RNA helicases have been shown to be hijacked by coronaviruses and to participate in essential DDX-mediated viral replication steps. Human DDX RNA helicases play essential roles in a broad array of biological processes and serve multiple roles at the virus-host interface. The viral proteins responsible for DDX interactions are highly conserved among coronaviruses, suggesting that they might also play conserved functions in the SARS-CoV-2 replication cycle. In this review, we provide an update of the structural and functional data of DDX as possible key factors involved in SARS-CoV-2 hijacking mechanisms. We also attempt to fill the existing gaps in the available structural information through homology modeling. Based on this information, we propose possible paths exploited by the virus to replicate more efficiently by taking advantage of host DDX proteins. As a general rule, sequestration of DDX helicases by SARS-CoV-2 is expected to play a pro-viral role in two ways: by enhancing key steps of the virus life cycle and, at the same time, by suppressing the host innate immune response.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
As intracellular parasites, viruses hijack the host cell metabolic machinery for their replication. Among other cellular proteins, the DEAD-box (DDX) RNA helicases have been shown to be hijacked by coronaviruses and to participate in essential DDX-mediated viral replication steps. Human DDX RNA helicases play essential roles in a broad array of biological processes and serve multiple roles at the virus-host interface. The viral proteins responsible for DDX interactions are highly conserved among coronaviruses, suggesting that they might also play conserved functions in the SARS-CoV-2 replication cycle. In this review, we provide an update of the structural and functional data of DDX as possible key factors involved in SARS-CoV-2 hijacking mechanisms. We also attempt to fill the existing gaps in the available structural information through homology modeling. Based on this information, we propose possible paths exploited by the virus to replicate more efficiently by taking advantage of host DDX proteins. As a general rule, sequestration of DDX helicases by SARS-CoV-2 is expected to play a pro-viral role in two ways: by enhancing key steps of the virus life cycle and, at the same time, by suppressing the host innate immune response. |
Brai A; Riva V; Saladini F; Zamperini C; Trivisani CI; Garbelli A; Pennisi C; Giannini A; Boccuto A; Bugli F; Martini M; Sanguinetti M; Zazzi M; Dreassi E; Botta M; Maga G DDX3X Inhibitors, an Effective Way to Overcome HIV-1 Resistance Targeting Host Proteins Journal Article In: European journal of medicinal chemistry, vol. 200, pp. 112319, 2020. @article{%a1:%Y_429,
title = {DDX3X Inhibitors, an Effective Way to Overcome HIV-1 Resistance Targeting Host Proteins},
author = {Brai A and Riva V and Saladini F and Zamperini C and Trivisani CI and Garbelli A and Pennisi C and Giannini A and Boccuto A and Bugli F and Martini M and Sanguinetti M and Zazzi M and Dreassi E and Botta M and Maga G},
url = {https://www.sciencedirect.com/science/article/pii/S0223523420302889},
doi = {10.1016/j.ejmech.2020.112319},
year = {2020},
date = {2020-01-01},
journal = {European journal of medicinal chemistry},
volume = {200},
pages = {112319},
abstract = {The huge resources that had gone into Human Immunodeficiency virus (HIV) research led to the development of potent antivirals able to suppress viral load in the majority of treated patients, thus dramatically increasing the life expectancy of people living with HIV. However, life-long treatments could result in the emergence of drug-resistant viruses that can progressively reduce the number of therapeutic options, facilitating the progression of the disease. In this scenario, we previously demonstrated that inhibitors of the human DDX3X helicase can represent an innovative approach for the simultaneous treatment of HIV and other viral infections such as Hepatitis c virus (HCV). We reported herein 6b, a novel DDX3X inhibitor that thanks to its distinct target of action is effective against HIV-1 strains resistant to currently approved drugs. Its improved in vitro ADME properties allowed us to perform preliminary in vivo studies in mice, which highlighted optimal biocompatibility and an improved bioavailability. These results represent a significant advancement in the development of DDX3X inhibitors as a novel class of broad spectrum and safe anti-HIV-1 drugs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The huge resources that had gone into Human Immunodeficiency virus (HIV) research led to the development of potent antivirals able to suppress viral load in the majority of treated patients, thus dramatically increasing the life expectancy of people living with HIV. However, life-long treatments could result in the emergence of drug-resistant viruses that can progressively reduce the number of therapeutic options, facilitating the progression of the disease. In this scenario, we previously demonstrated that inhibitors of the human DDX3X helicase can represent an innovative approach for the simultaneous treatment of HIV and other viral infections such as Hepatitis c virus (HCV). We reported herein 6b, a novel DDX3X inhibitor that thanks to its distinct target of action is effective against HIV-1 strains resistant to currently approved drugs. Its improved in vitro ADME properties allowed us to perform preliminary in vivo studies in mice, which highlighted optimal biocompatibility and an improved bioavailability. These results represent a significant advancement in the development of DDX3X inhibitors as a novel class of broad spectrum and safe anti-HIV-1 drugs. |
Brai A; Boccuto A; Monti M; Marchi S; Vicenti I; Saladini F; Trivisani CI; Pollutri A; Trombetta CM; Montomoli E; Riva V; Garbelli A; Nola EM; Zazzi M; Maga G; Dreassi E; Botta M Exploring the Implication of DDX3X in DENV Infection: Discovery of the First-in-Class DDX3X Fluorescent Inhibitor Journal Article In: ACS medicinal chemistry letters, vol. 11, no 5, pp. 956-962, 2020. @article{%a1:%Y_428,
title = {Exploring the Implication of DDX3X in DENV Infection: Discovery of the First-in-Class DDX3X Fluorescent Inhibitor},
author = {Brai A and Boccuto A and Monti M and Marchi S and Vicenti I and Saladini F and Trivisani CI and Pollutri A and Trombetta CM and Montomoli E and Riva V and Garbelli A and Nola EM and Zazzi M and Maga G and Dreassi E and Botta M},
url = {https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00681},
doi = {10.1021/acsmedchemlett.9b00681},
year = {2020},
date = {2020-01-01},
journal = {ACS medicinal chemistry letters},
volume = {11},
number = {5},
pages = {956-962},
abstract = {In the absence of effective drugs or vaccines for the treatment of the five Dengue Virus serotypes, the search for novel antiviral drugs is of primary importance for the scientific community. In this context, drug repurposing represents the most used strategy; however, the study of host targets is now attracting attention since it allows identification of broad-spectrum drugs endowed with high genetic barrier. In the last ten years our research group identified several small molecules DDX3X inhibitors and proved their efficacy against different viruses including novel emerging ones. Herein, starting from a screening of our compounds, we designed and synthesized novel derivatives with potent activity and high selectivity. Finally, we synthesized a fluorescent inhibitor that allowed us to study DDX3X cellular localization during DENV infection in vitro. Immunofluorescence analysis showed that our inhibitor colocalized with DDX3X, promoting the reduction of infected cells and recovering the number of viable cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In the absence of effective drugs or vaccines for the treatment of the five Dengue Virus serotypes, the search for novel antiviral drugs is of primary importance for the scientific community. In this context, drug repurposing represents the most used strategy; however, the study of host targets is now attracting attention since it allows identification of broad-spectrum drugs endowed with high genetic barrier. In the last ten years our research group identified several small molecules DDX3X inhibitors and proved their efficacy against different viruses including novel emerging ones. Herein, starting from a screening of our compounds, we designed and synthesized novel derivatives with potent activity and high selectivity. Finally, we synthesized a fluorescent inhibitor that allowed us to study DDX3X cellular localization during DENV infection in vitro. Immunofluorescence analysis showed that our inhibitor colocalized with DDX3X, promoting the reduction of infected cells and recovering the number of viable cells. |
Bono B; Franco G; Riva V; Garbelli A; Maga G Novel Insights into the Biochemical Mechanism of CK1epsilon and its Functional Interplay with DDX3X Journal Article In: International journal of molecular sciences, vol. 21, no 17, pp. E6449, 2020. @article{%a1:%Y_427,
title = {Novel Insights into the Biochemical Mechanism of CK1epsilon and its Functional Interplay with DDX3X},
author = {Bono B and Franco G and Riva V and Garbelli A and Maga G},
url = {https://www.mdpi.com/1422-0067/21/17/6449},
doi = {10.3390/ijms21176449},
year = {2020},
date = {2020-01-01},
journal = {International journal of molecular sciences},
volume = {21},
number = {17},
pages = {E6449},
abstract = {Casein Kinase 1 epsilon (CK1epsilon) is a member of the serine (Ser)/threonine (Thr) CK1 family, known to have crucial roles in several biological scenarios and, ever more frequently, in pathological contexts, such as cancer. Recently, the human DEAD-box RNA helicase 3 X-linked (DDX3X), involved in cancer proliferation and viral infections, has been identified as one of CK1epsilon substrates and its positive regulator in the Wnt/beta-catenin network. However, the way by which these two proteins influence each other has not been fully clarified. In order to further investigate their interplay, we defined the kinetic parameters of CK1epsilon towards its substrates: ATP, casein, Dvl2 and DDX3X. CK1epsilon affinity for ATP depends on the nature of the substrate: increasing of casein concentrations led to an increase of KmATP, while increasing DDX3X reduced it. In literature, DDX3X is described to act as an allosteric activator of CK1epsilon. However, when we performed kinase reactions combining DDX3X and casein, we did not find a positive effect of DDX3X on casein phosphorylation by CK1epsilon, while both substrates were phosphorylated in a competitive manner. Moreover, CK1epsilon positively stimulates DDX3X ATPase activity. Our data provide a more detailed kinetic characterization on the functional interplay of these two proteins.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Casein Kinase 1 epsilon (CK1epsilon) is a member of the serine (Ser)/threonine (Thr) CK1 family, known to have crucial roles in several biological scenarios and, ever more frequently, in pathological contexts, such as cancer. Recently, the human DEAD-box RNA helicase 3 X-linked (DDX3X), involved in cancer proliferation and viral infections, has been identified as one of CK1epsilon substrates and its positive regulator in the Wnt/beta-catenin network. However, the way by which these two proteins influence each other has not been fully clarified. In order to further investigate their interplay, we defined the kinetic parameters of CK1epsilon towards its substrates: ATP, casein, Dvl2 and DDX3X. CK1epsilon affinity for ATP depends on the nature of the substrate: increasing of casein concentrations led to an increase of KmATP, while increasing DDX3X reduced it. In literature, DDX3X is described to act as an allosteric activator of CK1epsilon. However, when we performed kinase reactions combining DDX3X and casein, we did not find a positive effect of DDX3X on casein phosphorylation by CK1epsilon, while both substrates were phosphorylated in a competitive manner. Moreover, CK1epsilon positively stimulates DDX3X ATPase activity. Our data provide a more detailed kinetic characterization on the functional interplay of these two proteins. |
Nalli M; Armijos Rivera JI; Masci D; Coluccia A; Badia R; Riveira-Munoz E; Brambilla A; Cinquina E; Turriziani O; Falasca F; Catalano M; Limatola C; Este JA; Maga G; Silvestri R; Crespan E; La Regina G New indolylarylsulfone non-nucleoside reverse transcriptase inhibitors show low nanomolar inhibition of single and double HIV-1 mutant strains Journal Article In: European journal of medicinal chemistry, vol. 208, pp. 112696, 2020. @article{%a1:%Y_460,
title = {New indolylarylsulfone non-nucleoside reverse transcriptase inhibitors show low nanomolar inhibition of single and double HIV-1 mutant strains},
author = {Nalli M and {Armijos Rivera JI} and Masci D and Coluccia A and Badia R and {Riveira-Munoz E} and Brambilla A and Cinquina E and Turriziani O and Falasca F and Catalano M and Limatola C and Este JA and Maga G and Silvestri R and Crespan E and {La Regina G}},
url = {https://www.sciencedirect.com/science/article/pii/S0223523420306681?via%3Dihub},
doi = {10.1016/j.ejmech.2020.112696},
year = {2020},
date = {2020-01-01},
journal = {European journal of medicinal chemistry},
volume = {208},
pages = {112696},
abstract = {We designed and synthesized 21 new indolylarylsulfones (IASs) as new HIV-1 NNRTIs. Among these, IAS 12 exhibited a remarkable antiviral activity against single and double mutants (K103N EC50 = <0.7 nM; Y181C EC50 = <0.7 nM; Y188L EC50 = 21.3 nM; K103N-Y181C EC50 = 6.2 nM), resulting equally or more active than previuosly reported IAS 6 and some approved anti-HIV-1 drugs. Docking and molecular dynamics simulations of compound 12 in complex with WT, Y181C, Y188L, K103N and K103N-Y181C RTs clarified a general binding mode that was consistent with biological results. Kinetic experiments disclosed that derivative 12 preferentially binds WT and K103N-Y181C RTs to binary and ternary complexes, respectively.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We designed and synthesized 21 new indolylarylsulfones (IASs) as new HIV-1 NNRTIs. Among these, IAS 12 exhibited a remarkable antiviral activity against single and double mutants (K103N EC50 = <0.7 nM; Y181C EC50 = <0.7 nM; Y188L EC50 = 21.3 nM; K103N-Y181C EC50 = 6.2 nM), resulting equally or more active than previuosly reported IAS 6 and some approved anti-HIV-1 drugs. Docking and molecular dynamics simulations of compound 12 in complex with WT, Y181C, Y188L, K103N and K103N-Y181C RTs clarified a general binding mode that was consistent with biological results. Kinetic experiments disclosed that derivative 12 preferentially binds WT and K103N-Y181C RTs to binary and ternary complexes, respectively. |
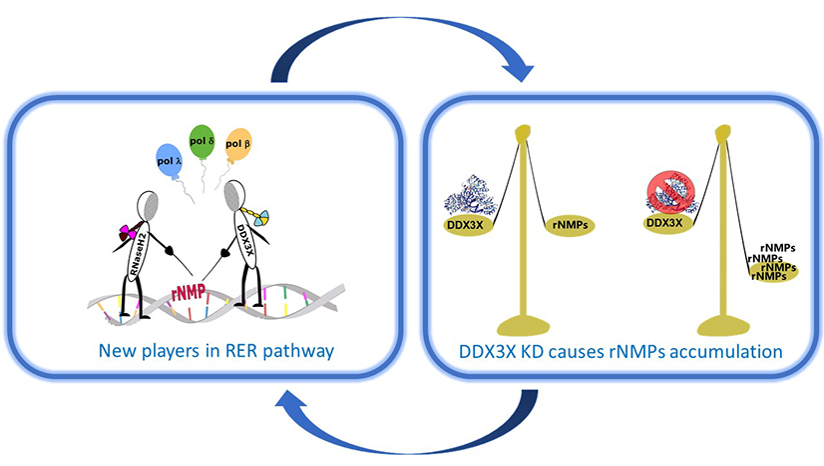
Riva V; Garbelli A; Casiraghi F; Arena F; Trivisani CI; Gagliardi A; Bini L; Schroeder M; Maffia A; Sabbioneda S; Maga G Novel alternative ribonucleotide excision repair pathways in human cells by DDX3X and specialized DNA polymerases. Journal Article In: Nucleic acids research, vol. 48, no 20, pp. 11551-11565, 2020. @article{%a1:%Y_472,
title = {Novel alternative ribonucleotide excision repair pathways in human cells by DDX3X and specialized DNA polymerases. },
author = {Riva V and Garbelli A and Casiraghi F and Arena F and Trivisani CI and Gagliardi A and Bini L and Schroeder M and Maffia A and Sabbioneda S and Maga G},
url = {https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7672437/},
doi = {10.1093/nar/gkaa948},
year = {2020},
date = {2020-01-01},
journal = {Nucleic acids research},
volume = {48},
number = {20},
pages = {11551-11565},
abstract = {Removal of ribonucleotides (rNMPs) incorporated into the genome by the ribonucleotide excision repair (RER) is essential to avoid genetic instability. In eukaryotes, the RNaseH2 is the only known enzyme able to incise 5' of the rNMP, starting the RER process, which is subsequently carried out by replicative DNA polymerases (Pols) delta or epsilon, together with Flap endonuclease 1 (Fen-1) and DNA ligase 1. Here, we show that the DEAD-box RNA helicase DDX3X has RNaseH2-like activity and can support fully reconstituted in vitro RER reactions, not only with Pol δ but also with the repair Pols beta and lambda. Silencing of DDX3X causes accumulation of rNMPs in the cellular genome. These results support the existence of alternative RER pathways conferring high flexibility to human cells in responding to the threat posed by rNMPs incorporation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Removal of ribonucleotides (rNMPs) incorporated into the genome by the ribonucleotide excision repair (RER) is essential to avoid genetic instability. In eukaryotes, the RNaseH2 is the only known enzyme able to incise 5' of the rNMP, starting the RER process, which is subsequently carried out by replicative DNA polymerases (Pols) delta or epsilon, together with Flap endonuclease 1 (Fen-1) and DNA ligase 1. Here, we show that the DEAD-box RNA helicase DDX3X has RNaseH2-like activity and can support fully reconstituted in vitro RER reactions, not only with Pol δ but also with the repair Pols beta and lambda. Silencing of DDX3X causes accumulation of rNMPs in the cellular genome. These results support the existence of alternative RER pathways conferring high flexibility to human cells in responding to the threat posed by rNMPs incorporation. |
Romano M; Ruggiero A; Squeglia F; Maga G; Berisio R A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping Journal Article In: Cells, vol. 9, no 5, pp. 1267, 2020. @article{%a1:%Y_473,
title = {A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping},
author = {Romano M and Ruggiero A and Squeglia F and Maga G and Berisio R},
url = {https://www.mdpi.com/2073-4409/9/5/1267},
doi = {10.3390/cells9051267},
year = {2020},
date = {2020-01-01},
journal = {Cells},
volume = {9},
number = {5},
pages = {1267},
abstract = {The current coronavirus disease-2019 (COVID-19) pandemic is due to the novel coronavirus SARS-CoV-2. The scientific community has mounted a strong response by accelerating research and innovation, and has quickly set the foundation for understanding the molecular determinants of the disease for the development of targeted therapeutic interventions. The replication of the viral genome within the infected cells is a key stage of the SARS-CoV-2 life cycle. It is a complex process involving the action of several viral and host proteins in order to perform RNA polymerization, proofreading and final capping. This review provides an update of the structural and functional data on the key actors of the replicatory machinery of SARS-CoV-2, to fill the gaps in the currently available structural data, which is mainly obtained through homology modeling. Moreover, learning from similar viruses, we collect data from the literature to reconstruct the pattern of interactions among the protein actors of the SARS-CoV-2 RNA polymerase machinery. Here, an important role is played by co-factors such as Nsp8 and Nsp10, not only as allosteric activators but also as molecular connectors that hold the entire machinery together to enhance the efficiency of RNA replication},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The current coronavirus disease-2019 (COVID-19) pandemic is due to the novel coronavirus SARS-CoV-2. The scientific community has mounted a strong response by accelerating research and innovation, and has quickly set the foundation for understanding the molecular determinants of the disease for the development of targeted therapeutic interventions. The replication of the viral genome within the infected cells is a key stage of the SARS-CoV-2 life cycle. It is a complex process involving the action of several viral and host proteins in order to perform RNA polymerization, proofreading and final capping. This review provides an update of the structural and functional data on the key actors of the replicatory machinery of SARS-CoV-2, to fill the gaps in the currently available structural data, which is mainly obtained through homology modeling. Moreover, learning from similar viruses, we collect data from the literature to reconstruct the pattern of interactions among the protein actors of the SARS-CoV-2 RNA polymerase machinery. Here, an important role is played by co-factors such as Nsp8 and Nsp10, not only as allosteric activators but also as molecular connectors that hold the entire machinery together to enhance the efficiency of RNA replication |
2019
|
Tassini S; Langron E; Delang L; Mirabelli C; Lanko K; Crespan E; Kissova M; Tagliavini G; Fontò G; Bertoni S; Palese S; Giorgio C; Ravanetti F; Ragionieri L; Zamperini C; Mancini A; Dreassi E; Maga G; Vergani P; Neyts J; Radi M Multitarget CFTR Modulators Endowed with Multiple Beneficial Side Effects for Cystic Fibrosis Patients: Toward a Simplified Therapeutic Approach Journal Article In: Journal of medicinal chemistry, vol. 62, no 23, pp. 10833-10847, 2019. @article{%a1:%Y_66,
title = {Multitarget CFTR Modulators Endowed with Multiple Beneficial Side Effects for Cystic Fibrosis Patients: Toward a Simplified Therapeutic Approach},
author = {Tassini S and Langron E and Delang L and Mirabelli C and Lanko K and Crespan E and Kissova M and Tagliavini G and Fontò G and Bertoni S and Palese S and Giorgio C and Ravanetti F and Ragionieri L and Zamperini C and Mancini A and Dreassi E and Maga G and Vergani P and Neyts J and Radi M},
url = {https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b01416},
doi = {10.1021/acs.jmedchem.9b01416},
year = {2019},
date = {2019-12-12},
journal = {Journal of medicinal chemistry},
volume = {62},
number = {23},
pages = {10833-10847},
abstract = {Cystic fibrosis (CF) is a multiorgan disease caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR). In addition to respiratory impairment due to mucus accumulation, viruses and bacteria trigger acute pulmonary exacerbations, accelerating disease progression and mortality rate. Treatment complexity increases with patients’ age, and simplifying the therapeutic regimen represents one of the key priorities in CF. We have recently reported the discovery of multitarget compounds able to “kill two birds with one stone” by targeting F508del-CFTR and PI4KIIIβ and thus acting simultaneously as CFTR correctors and broad-spectrum enterovirus (EV) inhibitors. Starting from these preliminary results, we report herein a hit-to-lead optimization and multidimensional structure–activity relationship (SAR) study that led to compound 23a. This compound showed good antiviral and F508del-CFTR correction potency, additivity/synergy with lumacaftor, and a promising in vitro absorption, distribution, metabolism, and excretion (ADME) profile. It was well tolerated in vivo with no sign of acute toxicity and histological alterations in key biodistribution organs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cystic fibrosis (CF) is a multiorgan disease caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR). In addition to respiratory impairment due to mucus accumulation, viruses and bacteria trigger acute pulmonary exacerbations, accelerating disease progression and mortality rate. Treatment complexity increases with patients’ age, and simplifying the therapeutic regimen represents one of the key priorities in CF. We have recently reported the discovery of multitarget compounds able to “kill two birds with one stone” by targeting F508del-CFTR and PI4KIIIβ and thus acting simultaneously as CFTR correctors and broad-spectrum enterovirus (EV) inhibitors. Starting from these preliminary results, we report herein a hit-to-lead optimization and multidimensional structure–activity relationship (SAR) study that led to compound 23a. This compound showed good antiviral and F508del-CFTR correction potency, additivity/synergy with lumacaftor, and a promising in vitro absorption, distribution, metabolism, and excretion (ADME) profile. It was well tolerated in vivo with no sign of acute toxicity and histological alterations in key biodistribution organs. |
Bavagnoli L; Campanini G; Forte M; Ceccotti G; Percivalle E; Bione S; Lisa A; Baldanti F; Maga G Identification of a novel antiviral micro-RNA targeting the NS1 protein of the H1N1 pandemic human influenza virus and a corresponding viral escape mutation. Journal Article In: Antiviral research, vol. 171, pp. 104593, 2019. @article{%a1:%Y%d,
title = {Identification of a novel antiviral micro-RNA targeting the NS1 protein of the H1N1 pandemic human influenza virus and a corresponding viral escape mutation.},
author = {Bavagnoli L and Campanini G and Forte M and Ceccotti G and Percivalle E and Bione S and Lisa A and Baldanti F and Maga G},
url = {https://www.sciencedirect.com/science/article/pii/S0166354219301640?via%3Dihub},
doi = {10.1016/j.antiviral.2019.104593},
year = {2019},
date = {2019-11-30},
journal = {Antiviral research},
volume = {171},
pages = {104593},
abstract = {The influenza A virus (IAV) NS1 protein is one of the major regulators of pathogenicity, being able to suppress innate immune response and host protein synthesis. In this study we identified the human micro RNA hsa-miR-1307-3p as a novel potent suppressor of NS1 expression and influenza virus replication. Transcriptomic analysis indicates that hsa-miR-1307-3p also negatively regulates apoptosis and promotes cell proliferation. In addition, we identified a novel mutation in the NS1 gene of A(H1N1)pdm09 strains circulating in Italy in the 2010-11 season, which enabled the virus to escape the hsa-miR-1307-3p inhibition, conferring replicative advantage to the virus in human cells. To the best of our knowledge, this is the first validation of suppression of IAV H1N1 NS1 by a human micro RNA and the first example of an escape mutation from micro RNA-mediated antiviral response for the A(H1N1)pdm09 virus.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The influenza A virus (IAV) NS1 protein is one of the major regulators of pathogenicity, being able to suppress innate immune response and host protein synthesis. In this study we identified the human micro RNA hsa-miR-1307-3p as a novel potent suppressor of NS1 expression and influenza virus replication. Transcriptomic analysis indicates that hsa-miR-1307-3p also negatively regulates apoptosis and promotes cell proliferation. In addition, we identified a novel mutation in the NS1 gene of A(H1N1)pdm09 strains circulating in Italy in the 2010-11 season, which enabled the virus to escape the hsa-miR-1307-3p inhibition, conferring replicative advantage to the virus in human cells. To the best of our knowledge, this is the first validation of suppression of IAV H1N1 NS1 by a human micro RNA and the first example of an escape mutation from micro RNA-mediated antiviral response for the A(H1N1)pdm09 virus. |
Brai A; Ronzini S; Riva V; Botta L; Zamperini C; Borgini M; Trivisani CI; Garbelli A; Pennisi C; Boccuto A; Saladini F; Zazzi M; Maga G; Botta M Synthesis and Antiviral Activity of Novel 1,3,4-Thiadiazole Inhibitors of DDX3X. Journal Article In: Molecules, vol. 24, no 21, pp. pii: E3988, 2019. @article{%a1:%Y%p,
title = {Synthesis and Antiviral Activity of Novel 1,3,4-Thiadiazole Inhibitors of DDX3X.},
author = {Brai A and Ronzini S and Riva V and Botta L and Zamperini C and Borgini M and Trivisani CI and Garbelli A and Pennisi C and Boccuto A and Saladini F and Zazzi M and Maga G and Botta M},
url = {https://www.mdpi.com/1420-3049/24/21/3988},
doi = {10.3390/molecules24213988},
year = {2019},
date = {2019-11-04},
journal = {Molecules},
volume = {24},
number = {21},
pages = { pii: E3988},
abstract = {The human ATPase/RNA helicase X-linked DEAD-box polypeptide 3 (DDX3X) emerged as a novel therapeutic target in the fight against both infectious diseases and cancer. Herein, a new family of DDX3X inhibitors was designed, synthesized, and tested for its inhibitory action on the ATPase activity of the enzyme. The potential use of the most promising derivatives it has been investigated by evaluating their anti-HIV-1 effects, revealing inhibitory activities in the low micromolar range. A preliminary ADME analysis demonstrated high metabolic stability and good aqueous solubility. The promising biological profile, together with the suitable in vitro pharmacokinetic properties, make these novel compounds a very good starting point for further development.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The human ATPase/RNA helicase X-linked DEAD-box polypeptide 3 (DDX3X) emerged as a novel therapeutic target in the fight against both infectious diseases and cancer. Herein, a new family of DDX3X inhibitors was designed, synthesized, and tested for its inhibitory action on the ATPase activity of the enzyme. The potential use of the most promising derivatives it has been investigated by evaluating their anti-HIV-1 effects, revealing inhibitory activities in the low micromolar range. A preliminary ADME analysis demonstrated high metabolic stability and good aqueous solubility. The promising biological profile, together with the suitable in vitro pharmacokinetic properties, make these novel compounds a very good starting point for further development. |
Fallacara AL; Passannanti R; Mori M; Iovenitti G; Musumeci F; Greco C; Crespan E; Kissova M; Maga G; Tarantelli C; Spriano F; Gaudio E; Bertoni F; Botta M; Schenone S Identification of a new family of pyrazolo[3,4-d]pyrimidine derivatives as multitarget Fyn-Blk-Lyn inhibitors active on B- and T-lymphoma cell lines. Journal Article In: European journal of medicinal chemistry, vol. 181, pp. 111545, 2019. @article{%a1:%Y%_34,
title = {Identification of a new family of pyrazolo[3,4-d]pyrimidine derivatives as multitarget Fyn-Blk-Lyn inhibitors active on B- and T-lymphoma cell lines.},
author = {Fallacara AL and Passannanti R and Mori M and Iovenitti G and Musumeci F and Greco C and Crespan E and Kissova M and Maga G and Tarantelli C and Spriano F and Gaudio E and Bertoni F and Botta M and Schenone S},
url = {https://www.sciencedirect.com/science/article/pii/S0223523419306695?via%3Dihub},
doi = {10.1016/j.ejmech.2019.07.048},
year = {2019},
date = {2019-11-01},
journal = {European journal of medicinal chemistry},
volume = {181},
pages = {111545},
abstract = {Abnormal activation of B-cell receptor (BCR) signaling plays a key role in the development of lymphoid malignancies, and could be reverted by the simultaneous inhibition of Lyn, Fyn and Blk, three members of the Src family kinase (SFK). Fyn and Blk are also promising targets for the treatment of some forms of T-cell non-Hodgkin lymphoma which point to the druggability of SFKs for the treatment of these cancers. We recently identified Si308 as a potent Fyn inhibitor, while preliminary data showed that it might also inhibit Lyn and Blk. Here, molecular modelling studies were coupled with enzymatic assays to further investigate the effect of Si308 on Lyn and Blk. A small library of pyrazolo[3,4-d]pyrimidines structurally related to Si308 was synthesized and tested on human lymphoma cell lines. Compound 2h emerged as a new multitarget inhibitor of Lyn, Fyn and Blk endowed with remarkable antiproliferative effects on human B and T lymphoma cell lines. Its favorable ADME properties make the compound suitable for further developments.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Abnormal activation of B-cell receptor (BCR) signaling plays a key role in the development of lymphoid malignancies, and could be reverted by the simultaneous inhibition of Lyn, Fyn and Blk, three members of the Src family kinase (SFK). Fyn and Blk are also promising targets for the treatment of some forms of T-cell non-Hodgkin lymphoma which point to the druggability of SFKs for the treatment of these cancers. We recently identified Si308 as a potent Fyn inhibitor, while preliminary data showed that it might also inhibit Lyn and Blk. Here, molecular modelling studies were coupled with enzymatic assays to further investigate the effect of Si308 on Lyn and Blk. A small library of pyrazolo[3,4-d]pyrimidines structurally related to Si308 was synthesized and tested on human lymphoma cell lines. Compound 2h emerged as a new multitarget inhibitor of Lyn, Fyn and Blk endowed with remarkable antiproliferative effects on human B and T lymphoma cell lines. Its favorable ADME properties make the compound suitable for further developments. |
Geronikaki A; Petrou A; Kartsev V; Eleftheriou P; Boga R; Bartolo B; Crespan E; Franco G; Maga G Molecular docking, design, synthesis and biological evaluation of novel 2,3-aryl-thiazolidin-4-ones as potent NNRTIs. Journal Article In: SAR and QSAR in environmental research, vol. 30, no 10, pp. 697-714, 2019. @article{%a1:%Y%_36,
title = {Molecular docking, design, synthesis and biological evaluation of novel 2,3-aryl-thiazolidin-4-ones as potent NNRTIs.},
author = {Geronikaki A and Petrou A and Kartsev V and Eleftheriou P and Boga R and Bartolo B and Crespan E and Franco G and Maga G},
doi = {10.1080/1062936X.2019.1653364.},
year = {2019},
date = {2019-10-31},
journal = {SAR and QSAR in environmental research},
volume = {30},
number = {10},
pages = {697-714},
abstract = {Nonnucleoside reverse transcriptase inhibitors (NNRTIs) remain the most promising anti-AIDS agents that target the HIV-1 reverse transcriptase enzyme (RT). However, the efficiency of approved NNRTI drugs has decreased by the appearance of drug-resistant viruses and side effects upon long-term usage. Thus, there is an urgent need for developing new, potent NNRTIs with broad spectrum against HIV-1 virus and with improved properties. In this study, a series of thiazolidinone derivatives was designed based on a butterfly mimicking scaffold consisting of a substituted benzothiazolyl moiety connected with a substituted phenyl ring via a thiazolidinone moiety. The most promising derivatives were selected using molecular docking analysis and PASS prediction program, synthesized and evaluated for HIV-1 RT inhibition. Five out of fifteen tested compounds exhibited good inhibitory action. It was observed that the presence of Cl or CN substituents at the position 6 of the benzothiazole ring in combination with two fluoro atoms at the ortho-positions or a hydrogen acceptor substituent at the 4-position of the phenyl ring are favourable for the HIV RT inhibitory activity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nonnucleoside reverse transcriptase inhibitors (NNRTIs) remain the most promising anti-AIDS agents that target the HIV-1 reverse transcriptase enzyme (RT). However, the efficiency of approved NNRTI drugs has decreased by the appearance of drug-resistant viruses and side effects upon long-term usage. Thus, there is an urgent need for developing new, potent NNRTIs with broad spectrum against HIV-1 virus and with improved properties. In this study, a series of thiazolidinone derivatives was designed based on a butterfly mimicking scaffold consisting of a substituted benzothiazolyl moiety connected with a substituted phenyl ring via a thiazolidinone moiety. The most promising derivatives were selected using molecular docking analysis and PASS prediction program, synthesized and evaluated for HIV-1 RT inhibition. Five out of fifteen tested compounds exhibited good inhibitory action. It was observed that the presence of Cl or CN substituents at the position 6 of the benzothiazole ring in combination with two fluoro atoms at the ortho-positions or a hydrogen acceptor substituent at the 4-position of the phenyl ring are favourable for the HIV RT inhibitory activity. |
Riva V; Maga G From the magic bullet to the magic target: exploiting the diverse roles of DDX3X in viral infections and tumorigenesis. Journal Article In: Future medicinal chemistry, vol. 11, no 11, pp. 1357-1381, 2019. @article{%a1:%Y_58,
title = {From the magic bullet to the magic target: exploiting the diverse roles of DDX3X in viral infections and tumorigenesis.},
author = {Riva V and Maga G},
url = {https://www.future-science.com/doi/full/10.4155/fmc-2018-0451?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dpubmed&},
doi = {10.4155/fmc-2018-0451},
year = {2019},
date = {2019-06-20},
journal = {Future medicinal chemistry},
volume = {11},
number = {11},
pages = {1357-1381},
abstract = {DDX3X is an ATPase/RNA helicase of the DEAD-box family and one of the most multifaceted helicases known up to date, acting in RNA metabolism, cell cycle control, apoptosis, stress response and innate immunity. Depending on the virus or the viral cycle stage, DDX3X can act either in a proviral fashion or as an antiviral factor. Similarly, in different cancer types, it can act either as an oncogene or a tumor-suppressor gene. Accumulating evidence indicated that DDX3X can be considered a promising target for anticancer and antiviral chemotherapy, but also that its exploitation requires a deeper understanding of the molecular mechanisms underlying its dual role in cancer and viral infections. In this review, we will summarize the known roles of DDX3X in different tumor types and viral infections, and the different inhibitors available, illustrating the possible advantages and potential caveats of their use as anticancer and antiviral drugs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
DDX3X is an ATPase/RNA helicase of the DEAD-box family and one of the most multifaceted helicases known up to date, acting in RNA metabolism, cell cycle control, apoptosis, stress response and innate immunity. Depending on the virus or the viral cycle stage, DDX3X can act either in a proviral fashion or as an antiviral factor. Similarly, in different cancer types, it can act either as an oncogene or a tumor-suppressor gene. Accumulating evidence indicated that DDX3X can be considered a promising target for anticancer and antiviral chemotherapy, but also that its exploitation requires a deeper understanding of the molecular mechanisms underlying its dual role in cancer and viral infections. In this review, we will summarize the known roles of DDX3X in different tumor types and viral infections, and the different inhibitors available, illustrating the possible advantages and potential caveats of their use as anticancer and antiviral drugs. |
Maga G Quando la cellula perde il controllo: capire il cancro per sconfiggerlo Book Zanichelli, 2019, ISBN: 9788808120472. @book{%a1:%Y_103,
title = {Quando la cellula perde il controllo: capire il cancro per sconfiggerlo},
author = {Maga G},
url = {https://www.zanichelli.it/ricerca/prodotti/quando-la-cellula-perde-il-controllo},
isbn = {9788808120472},
year = {2019},
date = {2019-03-15},
publisher = {Zanichelli},
abstract = {Con il metodo scientifico, non con le terapie alternative: solo così possiamo sconfiggere il cancro. Dalle frontiere della ricerca sono in arrivo farmaci a bersaglio molecolare, vaccini, immunoterapie e terapie geniche.
La trasformazione di una cellula sana in una cellula tumorale è irreversibile: una volta avviata, la cellula non torna più indietro. Grazie alle conoscenze sulle basi molecolari del cancro, oggi possiamo riconoscere le cellule tumorali, tracciarne l’identikit genetico e scegliere terapie personalizzate. Possiamo leggere il DNA alla ricerca delle mutazioni che provocano il cancro e sviluppare farmaci che colpiscono solo le cellule malate. Agiamo sul nostro sistema immunitario e lo rendiamo capace di riconoscere e aggredire i tumori. Arruoliamo virus e batteri per attaccare e uccidere le cellule cancerogene.
Le cure alternative sono invece una minaccia. Omeopatia, cristalloterapia, diete miracolose a base di cartilagine di squalo o bicarbonato sono trappole mortali. La ricerca scientifica è l’unico investimento sicuro per curare il cancro.},
keywords = {},
pubstate = {published},
tppubtype = {book}
}
Con il metodo scientifico, non con le terapie alternative: solo così possiamo sconfiggere il cancro. Dalle frontiere della ricerca sono in arrivo farmaci a bersaglio molecolare, vaccini, immunoterapie e terapie geniche.
La trasformazione di una cellula sana in una cellula tumorale è irreversibile: una volta avviata, la cellula non torna più indietro. Grazie alle conoscenze sulle basi molecolari del cancro, oggi possiamo riconoscere le cellule tumorali, tracciarne l’identikit genetico e scegliere terapie personalizzate. Possiamo leggere il DNA alla ricerca delle mutazioni che provocano il cancro e sviluppare farmaci che colpiscono solo le cellule malate. Agiamo sul nostro sistema immunitario e lo rendiamo capace di riconoscere e aggredire i tumori. Arruoliamo virus e batteri per attaccare e uccidere le cellule cancerogene.
Le cure alternative sono invece una minaccia. Omeopatia, cristalloterapia, diete miracolose a base di cartilagine di squalo o bicarbonato sono trappole mortali. La ricerca scientifica è l’unico investimento sicuro per curare il cancro. |
Brai A; Martelli F; Riva V; Garbelli A; Fazi R; Zamperini C; Pollutri A; Falsitta L; Ronzini S; Maccari L; Maga G; Giannecchini S; Botta M DDX3X Helicase Inhibitors as a New Strategy To Fight the West Nile Virus Infection. Journal Article In: Journal of medicinal chemistry, vol. 62, no 5, pp. 2333-2347, 2019. @article{%a1:%Y%o,
title = {DDX3X Helicase Inhibitors as a New Strategy To Fight the West Nile Virus Infection.},
author = {Brai A and Martelli F and Riva V and Garbelli A and Fazi R and Zamperini C and Pollutri A and Falsitta L and Ronzini S and Maccari L and Maga G and Giannecchini S and Botta M},
url = {https://pubs.acs.org/doi/10.1021/acs.jmedchem.8b01403},
doi = {10.1021/acs.jmedchem.8b01403},
year = {2019},
date = {2019-02-17},
journal = {Journal of medicinal chemistry},
volume = {62},
number = {5},
pages = {2333-2347},
abstract = {Increased frequency of arbovirus outbreaks in the last 10 years represents an important emergence for global health. Climate warming, extensive urbanization of tropical regions, and human migration flows facilitate the expansion of anthropophilic mosquitos and the emerging or re-emerging of new viral infections. Only recently the human adenosinetriphosphatase/RNA helicase X-linked DEAD-box polypeptide 3 (DDX3X) emerged as a novel therapeutic target in the fight against infectious diseases. Herein, starting from our previous studies, a new family of DDX3X inhibitors was designed, synthesized, validated on the target enzyme, and evaluated against the West Nile virus (WNV) infection. Time of addition experiments after virus infection indicated that the compounds exerted their antiviral activities after the entry process, likely at the protein translation step of WNV replication. Finally, the most interesting compounds were then analyzed for their in vitro pharmacokinetic parameters, revealing favorable absorption, distribution, metabolism, and excretion values. The good safety profile together with a good activity against WNV for which no treatments are currently available, make this new class of molecules a good starting point for further in vivo studies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Increased frequency of arbovirus outbreaks in the last 10 years represents an important emergence for global health. Climate warming, extensive urbanization of tropical regions, and human migration flows facilitate the expansion of anthropophilic mosquitos and the emerging or re-emerging of new viral infections. Only recently the human adenosinetriphosphatase/RNA helicase X-linked DEAD-box polypeptide 3 (DDX3X) emerged as a novel therapeutic target in the fight against infectious diseases. Herein, starting from our previous studies, a new family of DDX3X inhibitors was designed, synthesized, validated on the target enzyme, and evaluated against the West Nile virus (WNV) infection. Time of addition experiments after virus infection indicated that the compounds exerted their antiviral activities after the entry process, likely at the protein translation step of WNV replication. Finally, the most interesting compounds were then analyzed for their in vitro pharmacokinetic parameters, revealing favorable absorption, distribution, metabolism, and excretion values. The good safety profile together with a good activity against WNV for which no treatments are currently available, make this new class of molecules a good starting point for further in vivo studies. |
Nawrozkij M; Forgione M; Yablokov AS; Lucidi A; Tomaselli D; Patsilinakos A; Panella C; Hailu GS; Kirillov IA; Badia R; Riveira Muñoz E; Crespan E; Armijos-Rivera JI; Cirilli R; Ragno R; Este JA; Maga G; Mai A; Rotili D Effect of alpha-Methoxy Substitution on the anti-HIV Activity of Dihydropyrimidin-4(3H)-ones. Journal Article In: Journal of medicinal chemistry, vol. 62, no 2, pp. 604-621, 2019. @article{%a1:%Y_54,
title = {Effect of alpha-Methoxy Substitution on the anti-HIV Activity of Dihydropyrimidin-4(3H)-ones.},
author = {Nawrozkij M and Forgione M and Yablokov AS and Lucidi A and Tomaselli D and Patsilinakos A and Panella C and Hailu GS and Kirillov IA and Badia R and Riveira Muñoz E and Crespan E and Armijos-Rivera JI and Cirilli R and Ragno R and Este JA and Maga G and Mai A and Rotili D},
url = {https://pubs.acs.org/doi/10.1021/acs.jmedchem.8b01238},
doi = {10.1021/acs.jmedchem.8b01238},
year = {2019},
date = {2019-02-12},
journal = {Journal of medicinal chemistry},
volume = {62},
number = {2},
pages = {604-621},
abstract = {Conformational restriction applied to dihydrobenzylpyrimidin-4-(3H)-ones (DABOs) by the intoduction of a methyl group at the alpha-benzylic position is known to massively improve the anti-HIV-1 activity of these compounds. Here, we report the effects of methoxy substitution at the alpha-benzylic position in S-, NH-, and N,N-DABOs carrying 2,6-difluoro, 2-chloro-6-fluoro, or 2,6-dichloro substituted benzyl moieties. The various alpha-methoxy DABO series (12-14) present different SAR at the dihalo benzyl substitution, with the most potent compounds (12d,e and 13c) showing similar (picomolar/nanomolar) anti-HIV-1 potency as the corresponding alpha-methyl analogs against wt HIV-1, and 10- to 100-fold increased potency (up to low nanomolar) against clinically relevant K103N, Y181C, Y188L, IRLL98 and K103N+Y181C HIV-1 mutant strains, highlithing the importance of the alpha-methoxy substitution to provide highly efficient DABOs as "second generation" NNRTIs. HPLC enantioseparation of three of the most potent derivatives (12d, 13c and 14c) provided single enantiomers with significant enantioselectivity in HIV-1 inhibition. Computational studies allowed to correlate the best antiviral activity with the (R) absolute configuration at the alpha-methoxy stereogenic center.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Conformational restriction applied to dihydrobenzylpyrimidin-4-(3H)-ones (DABOs) by the intoduction of a methyl group at the alpha-benzylic position is known to massively improve the anti-HIV-1 activity of these compounds. Here, we report the effects of methoxy substitution at the alpha-benzylic position in S-, NH-, and N,N-DABOs carrying 2,6-difluoro, 2-chloro-6-fluoro, or 2,6-dichloro substituted benzyl moieties. The various alpha-methoxy DABO series (12-14) present different SAR at the dihalo benzyl substitution, with the most potent compounds (12d,e and 13c) showing similar (picomolar/nanomolar) anti-HIV-1 potency as the corresponding alpha-methyl analogs against wt HIV-1, and 10- to 100-fold increased potency (up to low nanomolar) against clinically relevant K103N, Y181C, Y188L, IRLL98 and K103N+Y181C HIV-1 mutant strains, highlithing the importance of the alpha-methoxy substitution to provide highly efficient DABOs as "second generation" NNRTIs. HPLC enantioseparation of three of the most potent derivatives (12d, 13c and 14c) provided single enantiomers with significant enantioselectivity in HIV-1 inhibition. Computational studies allowed to correlate the best antiviral activity with the (R) absolute configuration at the alpha-methoxy stereogenic center. |
2018
|
Kaptein SJF; Vincetti P; Crespan E; Rivera JIA; Costantino G; Maga G; Neyts J; Radi M Identification of Broad-Spectrum Dengue/Zika Virus Replication Inhibitors by Functionalization of Quinoline and 2,6-Diaminopurine Scaffolds. Journal Article In: ChemMedChem, vol. 13, no 14, pp. 1371-1376, 2018. @article{%a1:%Y_149,
title = {Identification of Broad-Spectrum Dengue/Zika Virus Replication Inhibitors by Functionalization of Quinoline and 2,6-Diaminopurine Scaffolds.},
author = {Kaptein SJF and Vincetti P and Crespan E and Rivera JIA and Costantino G and Maga G and Neyts J and Radi M},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/cmdc.201800178},
doi = {10.1002/cmdc.201800178},
year = {2018},
date = {2018-07-18},
journal = {ChemMedChem},
volume = {13},
number = {14},
pages = {1371-1376},
abstract = {Social and demographic changes across the world over the past 50 years have resulted in significant outbreaks of arboviruses such as dengue virus (DENV) and Zika virus (ZIKV). Despite the increased threat of infection, no approved drugs or fully protective vaccines are available to counteract the spread of DENV and ZIKV. The development of "broad-spectrum" antivirals (BSAs) that target common components of multiple viruses can be a more effective strategy to limit the rapid emergence of viral pathogens than the classic "one-bug/one-drug" approach. Starting from previously identified multitarget DENV inhibitors, herein we report the identification of novel 2,6-diaminopurine derivatives that are able to block the replication of both Zika virus and all serotypes of dengue virus (DENV 1-4) in infected cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Social and demographic changes across the world over the past 50 years have resulted in significant outbreaks of arboviruses such as dengue virus (DENV) and Zika virus (ZIKV). Despite the increased threat of infection, no approved drugs or fully protective vaccines are available to counteract the spread of DENV and ZIKV. The development of "broad-spectrum" antivirals (BSAs) that target common components of multiple viruses can be a more effective strategy to limit the rapid emergence of viral pathogens than the classic "one-bug/one-drug" approach. Starting from previously identified multitarget DENV inhibitors, herein we report the identification of novel 2,6-diaminopurine derivatives that are able to block the replication of both Zika virus and all serotypes of dengue virus (DENV 1-4) in infected cells. |
Molinari A; Fallacara AL; Di Maria S; Zamperini C; Poggialini F; Musumeci F; Schenone S; Angelucci A; Colapietro A; Crespan E; Kissova M; Maga G; Botta M Efficient optimization of pyrazolo[3,4-d]pyrimidines derivatives as c-Src kinase inhibitors in neuroblastoma treatment. Journal Article In: Bioorganic & medicinal chemistry letters, vol. 28, no 21, pp. 3454-3457, 2018. @article{%a1:%Y_162,
title = {Efficient optimization of pyrazolo[3,4-d]pyrimidines derivatives as c-Src kinase inhibitors in neuroblastoma treatment.},
author = {Molinari A and Fallacara AL and Di Maria S and Zamperini C and Poggialini F and Musumeci F and Schenone S and Angelucci A and Colapietro A and Crespan E and Kissova M and Maga G and Botta M},
url = {https://www.sciencedirect.com/science/article/pii/S0960894X18307558?via%3Dihub},
doi = {10.1016/j.bmcl.2018.09.024},
year = {2018},
date = {2018-02-17},
journal = {Bioorganic & medicinal chemistry letters},
volume = {28},
number = {21},
pages = {3454-3457},
abstract = {The proto-oncogene c-Src is a non-receptor tyrosine kinase which is involved in the regulation of many cellular processes, such as differentiation, adhesion and survival. c-Src hyperactivation has been detected in many tumors, including neuroblastoma (NB), one of the major causes of death from neoplasia in infancy. We already reported a large family of pyrazolo[3,4-d]pyrimidines active as c-Src inhibitors. Interestingly, some of these derivatives resulted also active on SH-SY5Y NB cell line. Herein, starting from our previous Free Energy Perturbation/Monte Carlo calculations, we report an optimization study which led to the identification of a new series of derivatives endowed with nanomolar Ki values against c-Src, interesting antiproliferative activity on SH-SY5Y cells and a suitable ADME profile.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The proto-oncogene c-Src is a non-receptor tyrosine kinase which is involved in the regulation of many cellular processes, such as differentiation, adhesion and survival. c-Src hyperactivation has been detected in many tumors, including neuroblastoma (NB), one of the major causes of death from neoplasia in infancy. We already reported a large family of pyrazolo[3,4-d]pyrimidines active as c-Src inhibitors. Interestingly, some of these derivatives resulted also active on SH-SY5Y NB cell line. Herein, starting from our previous Free Energy Perturbation/Monte Carlo calculations, we report an optimization study which led to the identification of a new series of derivatives endowed with nanomolar Ki values against c-Src, interesting antiproliferative activity on SH-SY5Y cells and a suitable ADME profile. |
2017
|
van Loon B; Hubscher U; Maga G Living on the Edge: DNA Polymerase Lambda between Genome Stability and Mutagenesis. Journal Article In: Chemical research in toxicology, vol. 30, no 11, pp. 1936-1941, 2017. @article{%a1:%Y_224,
title = {Living on the Edge: DNA Polymerase Lambda between Genome Stability and Mutagenesis.},
author = {van Loon B and Hubscher U and Maga G},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.chemrestox.7b00152},
doi = {10.1021/acs.chemrestox.7b00152},
year = {2017},
date = {2017-11-20},
journal = {Chemical research in toxicology},
volume = {30},
number = {11},
pages = {1936-1941},
abstract = {In human cells, only four DNA polymerases (pols) are necessary and sufficient for the duplication of the genetic information. However, more than a dozen DNA pols are required to maintain its integrity. Such a high degree of specialization makes DNA repair pols able to cope with specific lesions or repair pathways. On the other hand, the same DNA pols can have partially overlapping roles, which could result in possible conflicts of functions, if the DNA pols are not properly regulated. DNA pol λ is a typical example of such an enzyme. It is a multifunctional enzyme, endowed with special structural and biochemical properties, which make it capable of participating in different DNA repair pathways such as base excision repair, nonhomologous end joining, and translesion synthesis. However, when mutated or deregulated, DNA pol λ can also be a source of genetic instability. Its multiple roles in DNA damage tolerance and its ability in promoting tumor progression make it also a possible target for novel anticancer approaches.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In human cells, only four DNA polymerases (pols) are necessary and sufficient for the duplication of the genetic information. However, more than a dozen DNA pols are required to maintain its integrity. Such a high degree of specialization makes DNA repair pols able to cope with specific lesions or repair pathways. On the other hand, the same DNA pols can have partially overlapping roles, which could result in possible conflicts of functions, if the DNA pols are not properly regulated. DNA pol λ is a typical example of such an enzyme. It is a multifunctional enzyme, endowed with special structural and biochemical properties, which make it capable of participating in different DNA repair pathways such as base excision repair, nonhomologous end joining, and translesion synthesis. However, when mutated or deregulated, DNA pol λ can also be a source of genetic instability. Its multiple roles in DNA damage tolerance and its ability in promoting tumor progression make it also a possible target for novel anticancer approaches. |
Mentegari E; Crespan E; Bavagnoli L; Kissova M; Bertoletti F; Sabbioneda S; Imhof R; Sturla SJ; Nilforoushan A; Hubscher U; van Loon B; Maga G Ribonucleotide incorporation by human DNA polymerase eta impacts translesion synthesis and RNase H2 activity. Journal Article In: Nucleic Acids Research, vol. 45, no 5, pp. 2600-2614, 2017. @article{%a1:%Y_190,
title = {Ribonucleotide incorporation by human DNA polymerase eta impacts translesion synthesis and RNase H2 activity.},
author = {Mentegari E and Crespan E and Bavagnoli L and Kissova M and Bertoletti F and Sabbioneda S and Imhof R and Sturla SJ and Nilforoushan A and Hubscher U and van Loon B and Maga G},
url = {https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkw1275},
doi = {doi.org/10.1093/nar/gkw1275},
year = {2017},
date = {2017-03-16},
journal = {Nucleic Acids Research},
volume = {45},
number = {5},
pages = {2600-2614},
abstract = {Ribonucleotides (rNs) incorporated in the genome by DNA polymerases (Pols) are removed by RNase H2. Cytidine and guanosine preferentially accumulate over the other rNs. Here we show that human Pol η can incorporate cytidine monophosphate (rCMP) opposite guanine, 8-oxo-7,8-dihydroguanine, 8-methyl-2'-deoxyguanosine and a cisplatin intrastrand guanine crosslink (cis-PtGG), while it cannot bypass a 3-methylcytidine or an abasic site with rNs as substrates. Pol eta is also capable of synthesizing polyribonucleotide chains, and its activity is enhanced by its auxiliary factor DNA Pol delta interacting protein 2 (PolDIP2). Human RNase H2 removes cytidine and guanosine less efficiently than the other rNs and incorporation of rCMP opposite DNA lesions further reduces the efficiency of RNase H2. Experiments with XP-V cell extracts indicate Pol eta as the major basis of rCMP incorporation opposite cis-PtGG. These results suggest that translesion synthesis by Pol eta can contribute to the accumulation of rCMP in the genome, particularly opposite modified guanines.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ribonucleotides (rNs) incorporated in the genome by DNA polymerases (Pols) are removed by RNase H2. Cytidine and guanosine preferentially accumulate over the other rNs. Here we show that human Pol η can incorporate cytidine monophosphate (rCMP) opposite guanine, 8-oxo-7,8-dihydroguanine, 8-methyl-2'-deoxyguanosine and a cisplatin intrastrand guanine crosslink (cis-PtGG), while it cannot bypass a 3-methylcytidine or an abasic site with rNs as substrates. Pol eta is also capable of synthesizing polyribonucleotide chains, and its activity is enhanced by its auxiliary factor DNA Pol delta interacting protein 2 (PolDIP2). Human RNase H2 removes cytidine and guanosine less efficiently than the other rNs and incorporation of rCMP opposite DNA lesions further reduces the efficiency of RNase H2. Experiments with XP-V cell extracts indicate Pol eta as the major basis of rCMP incorporation opposite cis-PtGG. These results suggest that translesion synthesis by Pol eta can contribute to the accumulation of rCMP in the genome, particularly opposite modified guanines. |
Famiglini V; La Regina G; Coluccia A; Masci D; Brancale A; Badia R; Riveira-Munoz E; Esté JA; Crespan E; Brambilla A; Maga G; Catalano M; Limatola C; Formica FR; Cirilli R; Novellino E; Silvestri R Chiral Indolylarylsulfone Non-Nucleoside Reverse Transcriptase Inhibitors as New Potent and Broad Spectrum Anti-HIV-1 Agents. Journal Article In: Journal of medicinal chemistry, vol. 60, no 15, pp. 6528-6547, 2017. @article{%a1:%Y_198,
title = {Chiral Indolylarylsulfone Non-Nucleoside Reverse Transcriptase Inhibitors as New Potent and Broad Spectrum Anti-HIV-1 Agents.},
author = {Famiglini V and La Regina G and Coluccia A and Masci D and Brancale A and Badia R and Riveira-Munoz E and Esté JA and Crespan E and Brambilla A and Maga G and Catalano M and Limatola C and Formica FR and Cirilli R and Novellino E and Silvestri R},
url = {http://pubs.acs.org/doi/10.1021/acs.jmedchem.6b01906},
doi = {10.1021/acs.jmedchem.6b01906},
year = {2017},
date = {2017-02-23},
journal = {Journal of medicinal chemistry},
volume = {60},
number = {15},
pages = {6528-6547},
abstract = {We designed and synthesized a series of chiral indolyarylsulfones (IASs) as new HIV-1 NNRTIs. The new IASs 8-37 showed potent inhibition of the HIV-1 WT NL4-3 strain and of the mutant K103N, Y181C, Y188L, and K103N-Y181C HIV-1 strains. Six racemic mixtures, 8, 23-25, 31, and 33, were separated at semipreparative level into their pure enantiomers. The (R)-8 enantiomer bearing the chiral (α-methylbenzyl) was superior to the (S)-counterpart. IAS derivatives bearing the (S) alanine unit, (S)-23, (S,R)-25, (S)-31, and (S)-33, were remarkably more potent than the corresponding (R)-enantiomers. Compound 23 protected hippocampal neuronal cells from the excitotoxic insult, while efavirenz (EFV) did not contrast the neurotoxic effect of glutamate. The present results highlight the chiral IASs as new NNRTIs with improved resistance profile against the mutant HIV-1 strains and reduced neurotoxic effects.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We designed and synthesized a series of chiral indolyarylsulfones (IASs) as new HIV-1 NNRTIs. The new IASs 8-37 showed potent inhibition of the HIV-1 WT NL4-3 strain and of the mutant K103N, Y181C, Y188L, and K103N-Y181C HIV-1 strains. Six racemic mixtures, 8, 23-25, 31, and 33, were separated at semipreparative level into their pure enantiomers. The (R)-8 enantiomer bearing the chiral (α-methylbenzyl) was superior to the (S)-counterpart. IAS derivatives bearing the (S) alanine unit, (S)-23, (S,R)-25, (S)-31, and (S)-33, were remarkably more potent than the corresponding (R)-enantiomers. Compound 23 protected hippocampal neuronal cells from the excitotoxic insult, while efavirenz (EFV) did not contrast the neurotoxic effect of glutamate. The present results highlight the chiral IASs as new NNRTIs with improved resistance profile against the mutant HIV-1 strains and reduced neurotoxic effects. |
Garbelli A; Riva V; Crespan E; Maga G How to win the HIV-1 drug resistance hurdle race: running faster or jumping higher? Journal Article In: Biochemical journal, vol. 474, no 10, pp. 1559-1577, 2017. @article{%a1:%Y_219,
title = {How to win the HIV-1 drug resistance hurdle race: running faster or jumping higher?},
author = {Garbelli A and Riva V and Crespan E and Maga G},
url = {http://www.biochemj.org/content/474/10/1559.long},
doi = {10.1042/BCJ20160772},
year = {2017},
date = {2017-02-23},
journal = {Biochemical journal},
volume = {474},
number = {10},
pages = {1559-1577},
abstract = {Infections by the human immunodeficiency virus type 1 (HIV-1), the causative agent of the acquired immunodeficiency syndrome (AIDS), are still totaling an appalling 36.7 millions worldwide, with 1.1 million AIDS deaths/year and a similar number of yearly new infections. All this, in spite of the discovery of HIV-1 as the AIDS etiological agent more than 30 years ago and the introduction of an effective combinatorial antiretroviral therapy (cART), able to control disease progression, more than 20 years ago. Although very effective, current cART is plagued by the emergence of drug-resistant viral variants and most of the efforts in the development of novel direct-acting antiviral agents (DAAs) against HIV-1 have been devoted toward the fighting of resistance. In this review, rather than providing a detailed listing of all the drugs and the corresponding resistance mutations, we aim, through relevant examples, at presenting to the general reader the conceptual shift in the approaches that are being taken to overcome the viral resistance hurdle. From the classic 'running faster' strategy, based on the development of novel DAAs active against the mutant viruses selected by the previous drugs and/or presenting to the virus a high genetic barrier toward the development of resilience, to a 'jumping higher' approach, which looks at the cell, rather than the virus, as a source of valuable drug targets, in order to make the cellular environment non-permissive toward the replication of both wild-type and mutated viruses. 2017 The Author(s); published by Portland Press Limited on behalf of the Biochemical Society.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Infections by the human immunodeficiency virus type 1 (HIV-1), the causative agent of the acquired immunodeficiency syndrome (AIDS), are still totaling an appalling 36.7 millions worldwide, with 1.1 million AIDS deaths/year and a similar number of yearly new infections. All this, in spite of the discovery of HIV-1 as the AIDS etiological agent more than 30 years ago and the introduction of an effective combinatorial antiretroviral therapy (cART), able to control disease progression, more than 20 years ago. Although very effective, current cART is plagued by the emergence of drug-resistant viral variants and most of the efforts in the development of novel direct-acting antiviral agents (DAAs) against HIV-1 have been devoted toward the fighting of resistance. In this review, rather than providing a detailed listing of all the drugs and the corresponding resistance mutations, we aim, through relevant examples, at presenting to the general reader the conceptual shift in the approaches that are being taken to overcome the viral resistance hurdle. From the classic 'running faster' strategy, based on the development of novel DAAs active against the mutant viruses selected by the previous drugs and/or presenting to the virus a high genetic barrier toward the development of resilience, to a 'jumping higher' approach, which looks at the cell, rather than the virus, as a source of valuable drug targets, in order to make the cellular environment non-permissive toward the replication of both wild-type and mutated viruses. 2017 The Author(s); published by Portland Press Limited on behalf of the Biochemical Society. |
Tassini S; Sun L; Lanko K; Crespan E; Langron E; Falchi F; Kissova M; Armijos-Rivera JI; Delang L; Mirabelli C; Neyts J; Pieroni M; Cavalli A; Costantino G; Maga G; Vergani P; Leyssen P; Radi M Discovery of Multitarget Agents Active as Broad-Spectrum Antivirals and Correctors of Cystic Fibrosis Transmembrane Conductance Regulator for Associated Pulmonary Diseases. Journal Article In: Journal of medicinal chemistry, vol. 60, no 4, pp. 1400-1416, 2017. @article{%a1:%Y_199,
title = {Discovery of Multitarget Agents Active as Broad-Spectrum Antivirals and Correctors of Cystic Fibrosis Transmembrane Conductance Regulator for Associated Pulmonary Diseases.},
author = {Tassini S and Sun L and Lanko K and Crespan E and Langron E and Falchi F and Kissova M and Armijos-Rivera JI and Delang L and Mirabelli C and Neyts J and Pieroni M and Cavalli A and Costantino G and Maga G and Vergani P and Leyssen P and Radi M},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b01521},
doi = {10.1021/acs.jmedchem.6b01521},
year = {2017},
date = {2017-02-16},
journal = {Journal of medicinal chemistry},
volume = {60},
number = {4},
pages = {1400-1416},
abstract = {Enteroviruses (EVs) are among the most frequent infectious agents in humans worldwide and represent the leading cause of upper respiratory tract infections. No drugs for the treatment of EV infections are currently available. Recent studies have also linked EV infection with pulmonary exacerbations, especially in cystic fibrosis (CF) patients, and the importance of this link is probably underestimated. The aim of this work was to develop a new class of multitarget agents active both as broad-spectrum antivirals and as correctors of the F508del-cystic fibrosis transmembrane conductance regulator (CFTR) folding defect responsible for >90% of CF cases. We report herein the discovery of the first small molecules able to simultaneously act as correctors of the F508del-CFTR folding defect and as broad-spectrum antivirals against a panel of EVs representative of all major species.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Enteroviruses (EVs) are among the most frequent infectious agents in humans worldwide and represent the leading cause of upper respiratory tract infections. No drugs for the treatment of EV infections are currently available. Recent studies have also linked EV infection with pulmonary exacerbations, especially in cystic fibrosis (CF) patients, and the importance of this link is probably underestimated. The aim of this work was to develop a new class of multitarget agents active both as broad-spectrum antivirals and as correctors of the F508del-cystic fibrosis transmembrane conductance regulator (CFTR) folding defect responsible for >90% of CF cases. We report herein the discovery of the first small molecules able to simultaneously act as correctors of the F508del-CFTR folding defect and as broad-spectrum antivirals against a panel of EVs representative of all major species. |
Musumeci F; Fallacara AL; Brullo C; Grossi G; Botta L; Calandro P; Chiariello M; Kissova M; Crespan E; Maga G; Schenone S Identification of new pyrrolo[2,3-d]pyrimidines as Src tyrosine kinase inhibitors in vitro active against Glioblastoma. Journal Article In: European journal of medicinal chemistry, vol. 127, pp. 369-378, 2017. @article{%a1:%Y_209,
title = {Identification of new pyrrolo[2,3-d]pyrimidines as Src tyrosine kinase inhibitors in vitro active against Glioblastoma.},
author = {Musumeci F and Fallacara AL and Brullo C and Grossi G and Botta L and Calandro P and Chiariello M and Kissova M and Crespan E and Maga G and Schenone S},
url = {http://www.sciencedirect.com/science/article/pii/S0223523416310418},
doi = {dx.doi.org/10.1016/j.ejmech.2016.12.036},
year = {2017},
date = {2017-02-05},
journal = {European journal of medicinal chemistry},
volume = {127},
pages = {369-378},
abstract = {In the last few years, several pyrrolo-pyrimidine derivatives have been either approved by the US FDA and in other countries for the treatment of different diseases or are currently in phase I/II clinical trials. Herein we present the synthesis and the characterization of a novel series of pyrrolo[2,3-d]pyrimidines, compounds 8a-j, and their activity against Glioblastoma multiforme (GBM). Docking studies and MM-GBSA analysis revealed the ability of such compounds to efficiently interact with the ATP binding site of Src. Enzymatic assays against a mini-panel of kinases (Src, Fyn, EGFR, Kit, Flt3, Abl, AblT315I) have been performed, showing an unexpected selectivity of our pyrrolo[2,3-d]pyrimidines for Src. Finally, the derivatives were tested for their antiproliferative potency on U87 GBM cell line. Compound 8h showed a considerable cytotoxicity effect against U87 cell line with an IC50 value of 7.1 microM.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In the last few years, several pyrrolo-pyrimidine derivatives have been either approved by the US FDA and in other countries for the treatment of different diseases or are currently in phase I/II clinical trials. Herein we present the synthesis and the characterization of a novel series of pyrrolo[2,3-d]pyrimidines, compounds 8a-j, and their activity against Glioblastoma multiforme (GBM). Docking studies and MM-GBSA analysis revealed the ability of such compounds to efficiently interact with the ATP binding site of Src. Enzymatic assays against a mini-panel of kinases (Src, Fyn, EGFR, Kit, Flt3, Abl, AblT315I) have been performed, showing an unexpected selectivity of our pyrrolo[2,3-d]pyrimidines for Src. Finally, the derivatives were tested for their antiproliferative potency on U87 GBM cell line. Compound 8h showed a considerable cytotoxicity effect against U87 cell line with an IC50 value of 7.1 microM. |
2016
|
El-Moghazy SM; George RF; Osman EE; Elbatrawy AA; Kissova M; Colombo A; Crespan E; Maga G Novel pyrazolo[3,4-d]pyrimidines as dual Src-Abl inhibitors active against mutant form of Abl and the leukemia K-562 cell line. Journal Article In: European Journal of Medicinal Chemistry, vol. 123, pp. 1-13, 2016. @article{%a1:%Y_272,
title = {Novel pyrazolo[3,4-d]pyrimidines as dual Src-Abl inhibitors active against mutant form of Abl and the leukemia K-562 cell line.},
author = {El-Moghazy SM and George RF and Osman EE and Elbatrawy AA and Kissova M and Colombo A and Crespan E and Maga G},
url = {http://www.sciencedirect.com/science/article/pii/S0223523416305827},
doi = {10.1016/j.ejmech.2016.07.034},
year = {2016},
date = {2016-11-10},
journal = {European Journal of Medicinal Chemistry},
volume = {123},
pages = {1-13},
abstract = {Some novel 6-substituted pyrazolo[3,4-d]pyrimidines 4, 5, 6a-d, 7a-c, 8 and pyrazolo[4,3-e][1,2,4]triazolo[4,3-a]pyrimidines 9a-c, 10a-c, 11, 12a,b, 13a-c and 14 were synthesized and characterized by spectral and elemental analyses. They were screened for their biological activity in vitro against Abl and Src kinases. Compounds 7a and 7b revealed the highest activity against both wild and mutant Abl kinases as well as the Src kinase and the leukemia K-562 cell line. They can be considered as new hits for further structural optimization to obtain better activity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Some novel 6-substituted pyrazolo[3,4-d]pyrimidines 4, 5, 6a-d, 7a-c, 8 and pyrazolo[4,3-e][1,2,4]triazolo[4,3-a]pyrimidines 9a-c, 10a-c, 11, 12a,b, 13a-c and 14 were synthesized and characterized by spectral and elemental analyses. They were screened for their biological activity in vitro against Abl and Src kinases. Compounds 7a and 7b revealed the highest activity against both wild and mutant Abl kinases as well as the Src kinase and the leukemia K-562 cell line. They can be considered as new hits for further structural optimization to obtain better activity. |
Brai A; Fazi R; Tintori C; Zamperini C; Bugli F; Sanguinetti M; Stigliano E; Este' J; Badia R; Franco S; Martinez MA; Martinez JP; Meyerhans A; Saladini F; Zazzi M; Garbelli A; Maga G; Botta M Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Journal Article In: Proceedings of the National Academy of Sciences of the United States of America, vol. 113, no 9, pp. 5388-5393, 2016. @article{%a1:%Y_254,
title = {Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents.},
author = {Brai A and Fazi R and Tintori C and Zamperini C and Bugli F and Sanguinetti M and Stigliano E and Este' J and Badia R and Franco S and Martinez MA and Martinez JP and Meyerhans A and Saladini F and Zazzi M and Garbelli A and Maga G and Botta M},
url = {http://www.pnas.org/content/113/19/5388.long},
doi = {10.1073/pnas.1522987113},
year = {2016},
date = {2016-05-10},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {113},
number = {9},
pages = {5388-5393},
abstract = {Targeting a host factor essential for the replication of different viruses but not for the cells offers a higher genetic barrier to the development of resistance, may simplify therapy regimens for coinfections, and facilitates management of emerging viral diseases. DEAD-box polypeptide 3 (DDX3) is a human host factor required for the replication of several DNA and RNA viruses, including some of the most challenging human pathogens currently circulating, such as HIV-1, Hepatitis C virus, Dengue virus, and West Nile virus. Herein, we showed for the first time, to our knowledge, that the inhibition of DDX3 by a small molecule could be successfully exploited for the development of a broad spectrum antiviral agent. In addition to the multiple antiviral activities, hit compound 16d retained full activity against drug-resistant HIV-1 strains in the absence of cellular toxicity. Pharmacokinetics and toxicity studies in rats confirmed a good safety profile and bioavailability of 16d. Thus, DDX3 is here validated as a valuable therapeutic target.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Targeting a host factor essential for the replication of different viruses but not for the cells offers a higher genetic barrier to the development of resistance, may simplify therapy regimens for coinfections, and facilitates management of emerging viral diseases. DEAD-box polypeptide 3 (DDX3) is a human host factor required for the replication of several DNA and RNA viruses, including some of the most challenging human pathogens currently circulating, such as HIV-1, Hepatitis C virus, Dengue virus, and West Nile virus. Herein, we showed for the first time, to our knowledge, that the inhibition of DDX3 by a small molecule could be successfully exploited for the development of a broad spectrum antiviral agent. In addition to the multiple antiviral activities, hit compound 16d retained full activity against drug-resistant HIV-1 strains in the absence of cellular toxicity. Pharmacokinetics and toxicity studies in rats confirmed a good safety profile and bioavailability of 16d. Thus, DDX3 is here validated as a valuable therapeutic target. |
Kissova M; Maga G; Crespan E The human tyrosine kinase Kit and its gatekeeper mutant T670I, show different kinetic properties: Implications for drug design. Journal Article In: Bioorganic & Medicinal Chemistry, vol. 24, no 19, pp. 4555-4532, 2016. @article{%a1:%Y_321,
title = {The human tyrosine kinase Kit and its gatekeeper mutant T670I, show different kinetic properties: Implications for drug design.},
author = {Kissova M and Maga G and Crespan E},
url = {http://www.sciencedirect.com/science/article/pii/S0968089616305806},
doi = {10.1016/j.bmc.2016.07.059},
year = {2016},
date = {2016-03-10},
journal = {Bioorganic & Medicinal Chemistry},
volume = {24},
number = {19},
pages = {4555-4532},
abstract = {The tyrosine kinase Kit, a receptor for Stem Cell Factor, is involved, among others, in processes associated to cell survival, proliferation and migration. Upon physiological conditions, the activity of Kit is tightly regulated. However, primary mutations that lead to its constitutive activation are the causal oncogenic driver of gastrointestinal stromal tumours (GISTs). GISTs are known to be refractory to conventional therapies but the introduction of Imatinib, a selective inhibitor of tyrosine kinases Abl and Kit, significantly ameliorated the treatment options of GISTs patients. However, the acquisition of secondary mutations renders Kit resistant towards all available drugs. Mutation involving gatekeeper residues (such as V654a and T670I) influence both the structure and the catalytic activity of the enzyme. Therefore, detailed knowledge of the enzymatic properties of the mutant forms, in comparison with the wild type enzyme, is an important pre-requisite for the rational development of specific inhibitors. In this paper we report a thorough kinetic analysis of the reaction catalyzed by the Kit kinase and its gatekeeper mutated form T670I. Our results revealed the different mechanisms of action of these two enzymes and may open a new avenue for the future design of specific Kit inhibitors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The tyrosine kinase Kit, a receptor for Stem Cell Factor, is involved, among others, in processes associated to cell survival, proliferation and migration. Upon physiological conditions, the activity of Kit is tightly regulated. However, primary mutations that lead to its constitutive activation are the causal oncogenic driver of gastrointestinal stromal tumours (GISTs). GISTs are known to be refractory to conventional therapies but the introduction of Imatinib, a selective inhibitor of tyrosine kinases Abl and Kit, significantly ameliorated the treatment options of GISTs patients. However, the acquisition of secondary mutations renders Kit resistant towards all available drugs. Mutation involving gatekeeper residues (such as V654a and T670I) influence both the structure and the catalytic activity of the enzyme. Therefore, detailed knowledge of the enzymatic properties of the mutant forms, in comparison with the wild type enzyme, is an important pre-requisite for the rational development of specific inhibitors. In this paper we report a thorough kinetic analysis of the reaction catalyzed by the Kit kinase and its gatekeeper mutated form T670I. Our results revealed the different mechanisms of action of these two enzymes and may open a new avenue for the future design of specific Kit inhibitors. |
Crespan E; Furrer A; Rösinger M; Bertoletti F; Mentegari E; Chiapparini G; Imhof R; Ziegler N; Sturla SJ; Hubscher U; van Loon B; Maga G Impact of ribonucleotide incorporation by DNA polymerases beta and lambda on oxidative base excision repair. Journal Article In: Nature Communications, vol. 7, pp. 10805, 2016. @article{%a1:%Y_263,
title = {Impact of ribonucleotide incorporation by DNA polymerases beta and lambda on oxidative base excision repair.},
author = {Crespan E and Furrer A and Rösinger M and Bertoletti F and Mentegari E and Chiapparini G and Imhof R and Ziegler N and Sturla SJ and Hubscher U and van Loon B and Maga G},
url = {http://www.nature.com/ncomms/2016/160226/ncomms10805/full/ncomms10805.html},
doi = {10.1038/ncomms10805},
year = {2016},
date = {2016-02-26},
journal = {Nature Communications},
volume = {7},
pages = {10805},
abstract = {Oxidative stress is a very frequent source of DNA damage. Many cellular DNA polymerases (Pols) can incorporate ribonucleotides (rNMPs) during DNA synthesis. However, whether oxidative stress-triggered DNA repair synthesis contributes to genomic rNMPs incorporation is so far not fully understood. Human specialized Pols beta and lamdda are the important enzymes involved in the oxidative stress tolerance, acting both in base excision repair and in translesion synthesis past the very frequent oxidative lesion 7,8-dihydro-8-oxoguanine (8-oxo-G). We found that Pol beta, to a greater extent than Pol lambda can incorporate rNMPs opposite normal bases or 8-oxo-G, and with a different fidelity. Further, the incorporation of rNMPs opposite 8-oxo-G delays repair by DNA glycosylases. Studies in Pol beta- and lambda-deficient cell extracts suggest that Pol beta levels can greatly affect rNMP incorporation opposite oxidative DNA lesions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oxidative stress is a very frequent source of DNA damage. Many cellular DNA polymerases (Pols) can incorporate ribonucleotides (rNMPs) during DNA synthesis. However, whether oxidative stress-triggered DNA repair synthesis contributes to genomic rNMPs incorporation is so far not fully understood. Human specialized Pols beta and lamdda are the important enzymes involved in the oxidative stress tolerance, acting both in base excision repair and in translesion synthesis past the very frequent oxidative lesion 7,8-dihydro-8-oxoguanine (8-oxo-G). We found that Pol beta, to a greater extent than Pol lambda can incorporate rNMPs opposite normal bases or 8-oxo-G, and with a different fidelity. Further, the incorporation of rNMPs opposite 8-oxo-G delays repair by DNA glycosylases. Studies in Pol beta- and lambda-deficient cell extracts suggest that Pol beta levels can greatly affect rNMP incorporation opposite oxidative DNA lesions. |
Goodson WH 3rd; Lowe L; Carpenter DO; Gilbertson M; Manaf Ali A; Lopez de Cerain Salsamendi A; Lasfar A; Carnero A; Azqueta A; Amedei A; Charles AK; Collins AR; Ward A; Salzberg AC; Colacci A; Olsen AK; Berg A; Barclay BJ; Zhou BP; Blanco-Aparicio C; Baglole CJ; Dong C; Mondello C; Hsu CW; Naus CC; Yedjou C; Curran CS; Laird DW; Koch DC; Carlin DJ; Felsher DW; Roy D; Brown DG; Ratovitski E; Ryan EP; Corsini E; Rojas E; Moon EY; Laconi E; Marongiu F; Al-Mulla F; Chiaradonna F; Darroudi F; Martin FL; Van Schooten FJ; Goldberg GS; Wagemaker G; Nangami G; Calaf GM; Williams G; Wolf GT; Koppen G; Brunborg G; Kim Lyerly H; Krishnan H; Ab Hamid H; Yasaei H; Sone H; Kondoh H; Salem HK; Hsu HY; Park HH; Koturbash I; Miousse IR; Scovassi AI; Klaunig JE; Vondráček J; Raju J; Roman J; Wise JP Sr; Whitfield JR; Woodrick J; Christopher JA; Ochieng J; Martinez-Leal JF; Weisz J; Kravchenko J; Sun J; Prudhomme KR; Narayanan KB; Cohen-Solal KA; Moorwood K; Gonzalez L; Soucek L; Jian L; D'Abronzo LS; Lin LT; Li L; Gulliver L; McCawley LJ; Memeo L; Vermeulen L; Leyns L; Zhang L; Valverde M; Khatami M; Romano MF; Chapellier M; Williams MA; Wade M; Manjili MH; Lleonart M; Xia M; Gonzalez MJ; Karamouzis MV; Kirsch-Volders M; Vaccari M; Kuemmerle NB; Singh N; Cruickshanks N; Kleinstreuer N; van Larebeke N; Ahmed N; Ogunkua O; Krishnakumar PK; Vadgama P; Marignani PA; Ghosh PM; Ostrosky-Wegman P; Thompson P; Dent P; Heneberg P; Darbre P; Sing Leung P; Nangia-Makker P; Cheng QS; Robey RB; Al-Temaimi R; Roy R; Andrade-Vieira R; Sinha RK; Mehta R; Vento R; Di Fiore R; Ponce-Cusi R; Dornetshuber-Fleiss R; Nahta R; Castellino RC; Palorini R; Abd Hamid R; Langie SA; Eltom S; Brooks SA; Ryeom S; Wise SS; Bay SN; Harris SA; Papagerakis S; Romano S; Pavanello S; Eriksson S; Forte S; Casey SC; Luanpitpong S; Lee TJ; Otsuki T; Chen T; Massfelder T; Sanderson T; Guarnieri T; Hultman T; Dormoy V; Odero-Marah V; Sabbisetti V; Maguer-Satta V; Rathmell WK; Engström W; Decker WK; Bisson WH; Rojanasakul Y; Luqmani Y; Chen Z; Hu Z Llona-Minguez S; Hoglund A; Jacques SA; Johansson L; Calderon-Montano JM; Claesson M; Loseva O; Valerie NC; Lundbäck T; Piedrafita J; Maga G; Crespan E; Meijer L; Burgos Morón E; Baranczewski P; Hagbjork AL; Svensson R; Wiita E; Almlof I; Visnes T; Jeppsson F; Sigmundsson K; Jensen AJ; Artursson P; Jemth AS; Stenmark P; Warpman Berglund U; Scobie M; Helleday T Discovery of the First Potent and Selective Inhibitors of Human dCTP Pyrophosphatase 1. Journal Article In: Journal of medicinal chemistry, vol. 59, no 3, pp. 1140-1148, 2016. @article{%a1:%Y_292,
title = {Discovery of the First Potent and Selective Inhibitors of Human dCTP Pyrophosphatase 1.},
author = {{Goodson WH 3rd} and Lowe L and Carpenter DO and Gilbertson M and Manaf Ali A and Lopez de Cerain Salsamendi A and Lasfar A and Carnero A and Azqueta A and Amedei A and Charles AK and Collins AR and Ward A and Salzberg AC and Colacci A and Olsen AK and Berg A and Barclay BJ and Zhou BP and Blanco-Aparicio C and Baglole CJ and Dong C and Mondello C and Hsu CW and Naus CC and Yedjou C and Curran CS and Laird DW and Koch DC and Carlin DJ and Felsher DW and Roy D and Brown DG and Ratovitski E and Ryan EP and Corsini E and Rojas E and Moon EY and Laconi E and Marongiu F and Al-Mulla F and Chiaradonna F and Darroudi F and Martin FL and Van Schooten FJ and Goldberg GS and Wagemaker G and Nangami G and Calaf GM and Williams G and Wolf GT and Koppen G and Brunborg G and Kim Lyerly H and Krishnan H and Ab Hamid H and Yasaei H and Sone H and Kondoh H and Salem HK and Hsu HY and Park HH and Koturbash I and Miousse IR and Scovassi AI and Klaunig JE and Vondráček J and Raju J and Roman J and Wise JP Sr and Whitfield JR and Woodrick J and Christopher JA and Ochieng J and Martinez-Leal JF and Weisz J and Kravchenko J and Sun J and Prudhomme KR and Narayanan KB and Cohen-Solal KA and Moorwood K and Gonzalez L and Soucek L and Jian L and D'Abronzo LS and Lin LT and Li L and Gulliver L and McCawley LJ and Memeo L and Vermeulen L and Leyns L and Zhang L and Valverde M and Khatami M and Romano MF and Chapellier M and Williams MA and Wade M and Manjili MH and Lleonart M and Xia M and Gonzalez MJ and Karamouzis MV and Kirsch-Volders M and Vaccari M and Kuemmerle NB and Singh N and Cruickshanks N and Kleinstreuer N and van Larebeke N and Ahmed N and Ogunkua O and Krishnakumar PK and Vadgama P and Marignani PA and Ghosh PM and Ostrosky-Wegman P and Thompson P and Dent P and Heneberg P and Darbre P and Sing Leung P and Nangia-Makker P and Cheng QS and Robey RB and Al-Temaimi R and Roy R and Andrade-Vieira R and Sinha RK and Mehta R and Vento R and Di Fiore R and Ponce-Cusi R and Dornetshuber-Fleiss R and Nahta R and Castellino RC and Palorini R and Abd Hamid R and Langie SA and Eltom S and Brooks SA and Ryeom S and Wise SS and Bay SN and Harris SA and Papagerakis S and Romano S and Pavanello S and Eriksson S and Forte S and Casey SC and Luanpitpong S and Lee TJ and Otsuki T and Chen T and Massfelder T and Sanderson T and Guarnieri T and Hultman T and Dormoy V and Odero-Marah V and Sabbisetti V and Maguer-Satta V and Rathmell WK and Engström W and Decker WK and Bisson WH and Rojanasakul Y and Luqmani Y and Chen Z and Hu Z {Llona-Minguez S} and Hoglund A and Jacques SA and Johansson L and Calderon-Montano JM and Claesson M and Loseva O and Valerie NC and Lundbäck T and Piedrafita J and Maga G and Crespan E and Meijer L and Burgos Morón E and Baranczewski P and Hagbjork AL and Svensson R and Wiita E and Almlof I and Visnes T and Jeppsson F and Sigmundsson K and Jensen AJ and Artursson P and Jemth AS and Stenmark P and Warpman Berglund U and Scobie M and Helleday T},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01741},
doi = {10.1021/acs.jmedchem.5b01741},
year = {2016},
date = {2016-02-18},
journal = {Journal of medicinal chemistry},
volume = {59},
number = {3},
pages = {1140-1148},
abstract = {The dCTPase pyrophosphatase 1 (dCTPase) regulates the intracellular nucleotide pool through hydrolytic degradation of canonical and noncanonical nucleotide triphosphates (dNTPs). dCTPase is highly expressed in multiple carcinomas and is associated with cancer cell stemness. Here we report on the development of the first potent and selective dCTPase inhibitors that enhance the cytotoxic effect of cytidine analogues in leukemia cells. Boronate 30 displays a promising in vitro ADME profile, including plasma and mouse microsomal half-lives, aqueous solubility, cell permeability and CYP inhibition, deeming it a suitable compound for in vivo studies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The dCTPase pyrophosphatase 1 (dCTPase) regulates the intracellular nucleotide pool through hydrolytic degradation of canonical and noncanonical nucleotide triphosphates (dNTPs). dCTPase is highly expressed in multiple carcinomas and is associated with cancer cell stemness. Here we report on the development of the first potent and selective dCTPase inhibitors that enhance the cytotoxic effect of cytidine analogues in leukemia cells. Boronate 30 displays a promising in vitro ADME profile, including plasma and mouse microsomal half-lives, aqueous solubility, cell permeability and CYP inhibition, deeming it a suitable compound for in vivo studies. |
Radi M; Schneider R; Fallacara AL; Botta L; Crespan E; Tintori C; Maga G; Kissova M; Calgani A; Richters A; Musumeci F; Rauh D; Schenone S A cascade screening approach for the identification of Bcr-Abl myristate pocket binders active against wild type and T315I mutant. Journal Article In: Bioorganic & Medicinal Chemistry Letters, vol. 26, no 15, pp. 3436-3440, 2016. @article{%a1:%Y_304,
title = {A cascade screening approach for the identification of Bcr-Abl myristate pocket binders active against wild type and T315I mutant.},
author = {Radi M and Schneider R and Fallacara AL and Botta L and Crespan E and Tintori C and Maga G and Kissova M and Calgani A and Richters A and Musumeci F and Rauh D and Schenone S},
url = {http://www.sciencedirect.com/science/article/pii/S0960894X1630662X},
doi = {10.1016/j.bmcl.2016.06.051},
year = {2016},
date = {2016-02-17},
journal = {Bioorganic & Medicinal Chemistry Letters},
volume = {26},
number = {15},
pages = {3436-3440},
abstract = {The major clinical challenge in drug-resistant chronic myelogenous leukemia (CML) is currently represented by the Bcr-Abl T315I mutant, which is unresponsive to treatment with common first and second generation ATP-competitive tyrosine kinase inhibitors (TKIs). Allosteric inhibition of Bcr-Abl represent a new frontier in the fight against resistant leukemia and few candidates have been identified in the last few years. Among these, myristate pocket (MP) binders discovered by Novartis (e.g. GNF2/5) showed promising results, although they proved to be active against the T315I mutant only in combination with first and second generation ATP-competitive inhibitors. Here we used a cascade screening approach based on sequential fluorescence polarization (FP) screening, in silico docking/dynamics studies and kinetic-enzymatic studies to identify novel MP binders. A pyrazolo[3,4-d]pyrimidine derivative (6) has been identified as a promising allosteric inhibitor active on 32D leukemia cell lines (expressing Bcr-Abl WT and T315I) with no need of combination with any ATP-competitive inhibitor.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The major clinical challenge in drug-resistant chronic myelogenous leukemia (CML) is currently represented by the Bcr-Abl T315I mutant, which is unresponsive to treatment with common first and second generation ATP-competitive tyrosine kinase inhibitors (TKIs). Allosteric inhibition of Bcr-Abl represent a new frontier in the fight against resistant leukemia and few candidates have been identified in the last few years. Among these, myristate pocket (MP) binders discovered by Novartis (e.g. GNF2/5) showed promising results, although they proved to be active against the T315I mutant only in combination with first and second generation ATP-competitive inhibitors. Here we used a cascade screening approach based on sequential fluorescence polarization (FP) screening, in silico docking/dynamics studies and kinetic-enzymatic studies to identify novel MP binders. A pyrazolo[3,4-d]pyrimidine derivative (6) has been identified as a promising allosteric inhibitor active on 32D leukemia cell lines (expressing Bcr-Abl WT and T315I) with no need of combination with any ATP-competitive inhibitor. |
Mentegari E; Kissova M; Bavagnoli L; Maga G; Crespan E DNA Polymerases lambda and beta: The Double-Edged Swords of DNA Repair. Journal Article In: Genes (Basel), vol. 7, no 9, pp. e57, 2016. @article{%a1:%Y_299,
title = {DNA Polymerases lambda and beta: The Double-Edged Swords of DNA Repair.},
author = {Mentegari E and Kissova M and Bavagnoli L and Maga G and Crespan E},
url = {http://www.mdpi.com/2073-4425/7/9/57},
doi = {10.3390/genes7090057},
year = {2016},
date = {2016-02-11},
journal = {Genes (Basel)},
volume = {7},
number = {9},
pages = {e57},
abstract = {DNA is constantly exposed to both endogenous and exogenous damages. More than 10,000 DNA modifications are induced every day in each cell's genome. Maintenance of the integrity of the genome is accomplished by several DNA repair systems. The core enzymes for these pathways are the DNA polymerases. Out of 17 DNA polymerases present in a mammalian cell, at least 13 are specifically devoted to DNA repair and are often acting in different pathways. DNA polymerases beta and lambda are involved in base excision repair of modified DNA bases and translesion synthesis past DNA lesions. Polymerase lambda also participates in non-homologous end joining of DNA double-strand breaks. However, recent data have revealed that, depending on their relative levels, the cell cycle phase, the ratio between deoxy- and ribo-nucleotide pools and the interaction with particular auxiliary proteins, the repair reactions carried out by these enzymes can be an important source of genetic instability, owing to repair mistakes. This review summarizes the most recent results on the ambivalent properties of these enzymes in limiting or promoting genetic instability in mammalian cells, as well as their potential use as targets for anticancer chemotherapy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
DNA is constantly exposed to both endogenous and exogenous damages. More than 10,000 DNA modifications are induced every day in each cell's genome. Maintenance of the integrity of the genome is accomplished by several DNA repair systems. The core enzymes for these pathways are the DNA polymerases. Out of 17 DNA polymerases present in a mammalian cell, at least 13 are specifically devoted to DNA repair and are often acting in different pathways. DNA polymerases beta and lambda are involved in base excision repair of modified DNA bases and translesion synthesis past DNA lesions. Polymerase lambda also participates in non-homologous end joining of DNA double-strand breaks. However, recent data have revealed that, depending on their relative levels, the cell cycle phase, the ratio between deoxy- and ribo-nucleotide pools and the interaction with particular auxiliary proteins, the repair reactions carried out by these enzymes can be an important source of genetic instability, owing to repair mistakes. This review summarizes the most recent results on the ambivalent properties of these enzymes in limiting or promoting genetic instability in mammalian cells, as well as their potential use as targets for anticancer chemotherapy. |
Tintori C; Brai A; Dasso Lang MC; Deodato D; Greco AM; Bizzarri BM; Cascone L; Casian A; Zamperini C; Dreassi E; Crespan E; Maga G; Vanham G; Ceresola E; Canducci F; Arien KK; Botta M Development and in Vitro Evaluation of a Microbicide Gel Formulation for a Novel Non-Nucleoside Reverse Transcriptase Inhibitor Belonging to the N-Dihydroalkyloxybenzyloxopyrimidines (N-DABOs) Family. Journal Article In: Journal of medicinal chemistry, vol. 59, no 6, pp. 2747-2759, 2016. @article{%a1:%Y_314,
title = {Development and in Vitro Evaluation of a Microbicide Gel Formulation for a Novel Non-Nucleoside Reverse Transcriptase Inhibitor Belonging to the N-Dihydroalkyloxybenzyloxopyrimidines (N-DABOs) Family.},
author = {Tintori C and Brai A and Dasso Lang MC and Deodato D and Greco AM and Bizzarri BM and Cascone L and Casian A and Zamperini C and Dreassi E and Crespan E and Maga G and Vanham G and Ceresola E and Canducci F and Arien KK and Botta M},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01979},
doi = {10.1021/acs.jmedchem.5b01979},
year = {2016},
date = {2016-02-10},
journal = {Journal of medicinal chemistry},
volume = {59},
number = {6},
pages = {2747-2759},
abstract = {Preventing HIV transmission by the use of a vaginal microbicide is a topic of considerable interest in the fight against AIDS. Both a potent anti-HIV agent and an efficient formulation are required to develop a successful microbicide. In this regard, molecules able to inhibit the HIV replication before the integration of the viral DNA into the genetic material of the host cells, such as entry inhibitors or reverse transcriptase inhibitors (RTIs), are ideal candidates for prevention purpose. Among RTIs, S- and N-dihydroalkyloxybenzyloxopyrimidines (S-DABOs and N-DABOs) are interesting compounds active at nanomolar concentration against wild type of RT and with a very interesting activity against RT mutations. Herein, novel N-DABOs were synthesized and tested as anti-HIV agents. Furthermore, their mode of binding was studied by molecular modeling. At the same time, a vaginal microbicide gel formulation was developed and tested for one of the most promising candidates.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Preventing HIV transmission by the use of a vaginal microbicide is a topic of considerable interest in the fight against AIDS. Both a potent anti-HIV agent and an efficient formulation are required to develop a successful microbicide. In this regard, molecules able to inhibit the HIV replication before the integration of the viral DNA into the genetic material of the host cells, such as entry inhibitors or reverse transcriptase inhibitors (RTIs), are ideal candidates for prevention purpose. Among RTIs, S- and N-dihydroalkyloxybenzyloxopyrimidines (S-DABOs and N-DABOs) are interesting compounds active at nanomolar concentration against wild type of RT and with a very interesting activity against RT mutations. Herein, novel N-DABOs were synthesized and tested as anti-HIV agents. Furthermore, their mode of binding was studied by molecular modeling. At the same time, a vaginal microbicide gel formulation was developed and tested for one of the most promising candidates. |
Maga G Batteri Spazzini e Virus che curano: come le biotecnologie riscrivono la vita Book Zanichelli, Bologna, 2016, ISBN: 9788808920836. @book{CNRPRODOTTI368292,
title = {Batteri Spazzini e Virus che curano: come le biotecnologie riscrivono la vita},
author = {Maga G},
url = {http://www.zanichelli.it/ricerca/prodotti/batteri-spazzini-e-virus-che-curano},
isbn = {9788808920836},
year = {2016},
date = {2016-01-01},
publisher = {Zanichelli},
address = {Bologna},
keywords = {},
pubstate = {published},
tppubtype = {book}
}
|
2015
|
Fazi R; Tintori C; Brai A; Botta L; Selvaraj M; Garbelli A; Maga G; Botta M Homology Model-Based Virtual Screening for the Identification of Human Helicase DDX3 Inhibitors. Journal Article In: Journal of Chemical Information and Modeling, vol. 55, no 11, pp. 2443-2454, 2015. @article{%a1:%Y_370,
title = {Homology Model-Based Virtual Screening for the Identification of Human Helicase DDX3 Inhibitors.},
author = {Fazi R and Tintori C and Brai A and Botta L and Selvaraj M and Garbelli A and Maga G and Botta M},
url = {https://pubs.acs.org/doi/10.1021/acs.jcim.5b00419},
doi = {10.1021/acs.jcim.5b00419},
year = {2015},
date = {2015-11-23},
journal = {Journal of Chemical Information and Modeling},
volume = {55},
number = {11},
pages = {2443-2454},
abstract = {Targeting cellular cofactors instead of viral enzymes represents a new strategy to combat infectious diseases, which should help to overcome the problem of viral resistance. Recently, it has been revealed that the cellular ATPase/RNA helicase X-linked DEAD-box polypeptide 3 (DDX3) is an essential host factor for the replication of several viruses such as HIV, HCV, JEV, Dengue, and West Nile. Accordingly, a drug targeting DDX3 could theoretically inhibit all viruses that are dependent on this host factor. Herein, for the first time, a model of hDDX3 in its closed conformation, which binds the viral RNA was developed by using the homology module of Prime through the Maestro interface of Schrodinger. Next, a structure-based virtual screening protocol was applied to identify DDX3 small molecule inhibitors targeting the RNA binding pocket. As a result, an impressive hit rate of 40% was obtained with the identification of 10 active compounds out of the 25 tested small molecules. The best poses of the active ligands highlighted the crucial residues to be targeted for the inhibition of the helicase activity of DDX3. The obtained results confirm the reliability of the constructed DDX3/RNA model and the proposed computational strategy for investigating novel DDX3 inhibitors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Targeting cellular cofactors instead of viral enzymes represents a new strategy to combat infectious diseases, which should help to overcome the problem of viral resistance. Recently, it has been revealed that the cellular ATPase/RNA helicase X-linked DEAD-box polypeptide 3 (DDX3) is an essential host factor for the replication of several viruses such as HIV, HCV, JEV, Dengue, and West Nile. Accordingly, a drug targeting DDX3 could theoretically inhibit all viruses that are dependent on this host factor. Herein, for the first time, a model of hDDX3 in its closed conformation, which binds the viral RNA was developed by using the homology module of Prime through the Maestro interface of Schrodinger. Next, a structure-based virtual screening protocol was applied to identify DDX3 small molecule inhibitors targeting the RNA binding pocket. As a result, an impressive hit rate of 40% was obtained with the identification of 10 active compounds out of the 25 tested small molecules. The best poses of the active ligands highlighted the crucial residues to be targeted for the inhibition of the helicase activity of DDX3. The obtained results confirm the reliability of the constructed DDX3/RNA model and the proposed computational strategy for investigating novel DDX3 inhibitors. |
Bavagnoli L; Cucuzza S; Campanini G; Rovida F; Paolucci S; Baldanti F; Maga G The novel influenza A virus protein PA-X and its naturally deleted variant show different enzymatic properties in comparison to the viral endonuclease PA. Journal Article In: Nucleic Acids Research, vol. 43, no 19, pp. 9405-9417, 2015. @article{%a1:%Y_414,
title = {The novel influenza A virus protein PA-X and its naturally deleted variant show different enzymatic properties in comparison to the viral endonuclease PA.},
author = {Bavagnoli L and Cucuzza S and Campanini G and Rovida F and Paolucci S and Baldanti F and Maga G},
url = {https://academic.oup.com/nar/article/43/19/9405/2528196},
doi = {10.1093/nar/gkv926},
year = {2015},
date = {2015-10-15},
journal = {Nucleic Acids Research},
volume = {43},
number = {19},
pages = {9405-9417},
abstract = {The PA protein of Influenza A virus (IAV) encoded by segment 3 acts as a specialized RNA endonuclease in the transcription of the viral genome. The same genomic segment encodes for a second shorter protein, termed PA-X, with the first 191 N-terminal aminoacids (aa) identical to PA, but with a completely different C-ter domain of 61 aa, due to a ribosomal frameshifting. In addition, it has been shown that several IAV isolates encode for a naturally truncated PA-X variant, PAXΔC20, missing the last 20 aa. The biochemical properties of PA-X and PAXΔC20 have been poorly investigated so far. Here, we have carried out an enzymatic characterization of PA-X and its naturally deleted form, in comparison with PA from the human IAV strain A/WSN/33 (H1N1). Our results showed, to the best of our knowledge for the first time, that PA-X possesses an endonucleolytic activity. Both PA and PA-X preferentially cut single stranded RNA regions, but with some differences. In addition, we showed that PAXΔC20 has severely reduced nuclease activity. These results point to a previously undetected role of the last C-ter 20 aa for the catalytic activity of PA-X and support distinct roles for these proteins in the viral life cycle. The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The PA protein of Influenza A virus (IAV) encoded by segment 3 acts as a specialized RNA endonuclease in the transcription of the viral genome. The same genomic segment encodes for a second shorter protein, termed PA-X, with the first 191 N-terminal aminoacids (aa) identical to PA, but with a completely different C-ter domain of 61 aa, due to a ribosomal frameshifting. In addition, it has been shown that several IAV isolates encode for a naturally truncated PA-X variant, PAXΔC20, missing the last 20 aa. The biochemical properties of PA-X and PAXΔC20 have been poorly investigated so far. Here, we have carried out an enzymatic characterization of PA-X and its naturally deleted form, in comparison with PA from the human IAV strain A/WSN/33 (H1N1). Our results showed, to the best of our knowledge for the first time, that PA-X possesses an endonucleolytic activity. Both PA and PA-X preferentially cut single stranded RNA regions, but with some differences. In addition, we showed that PAXΔC20 has severely reduced nuclease activity. These results point to a previously undetected role of the last C-ter 20 aa for the catalytic activity of PA-X and support distinct roles for these proteins in the viral life cycle. The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research. |