RICERCA
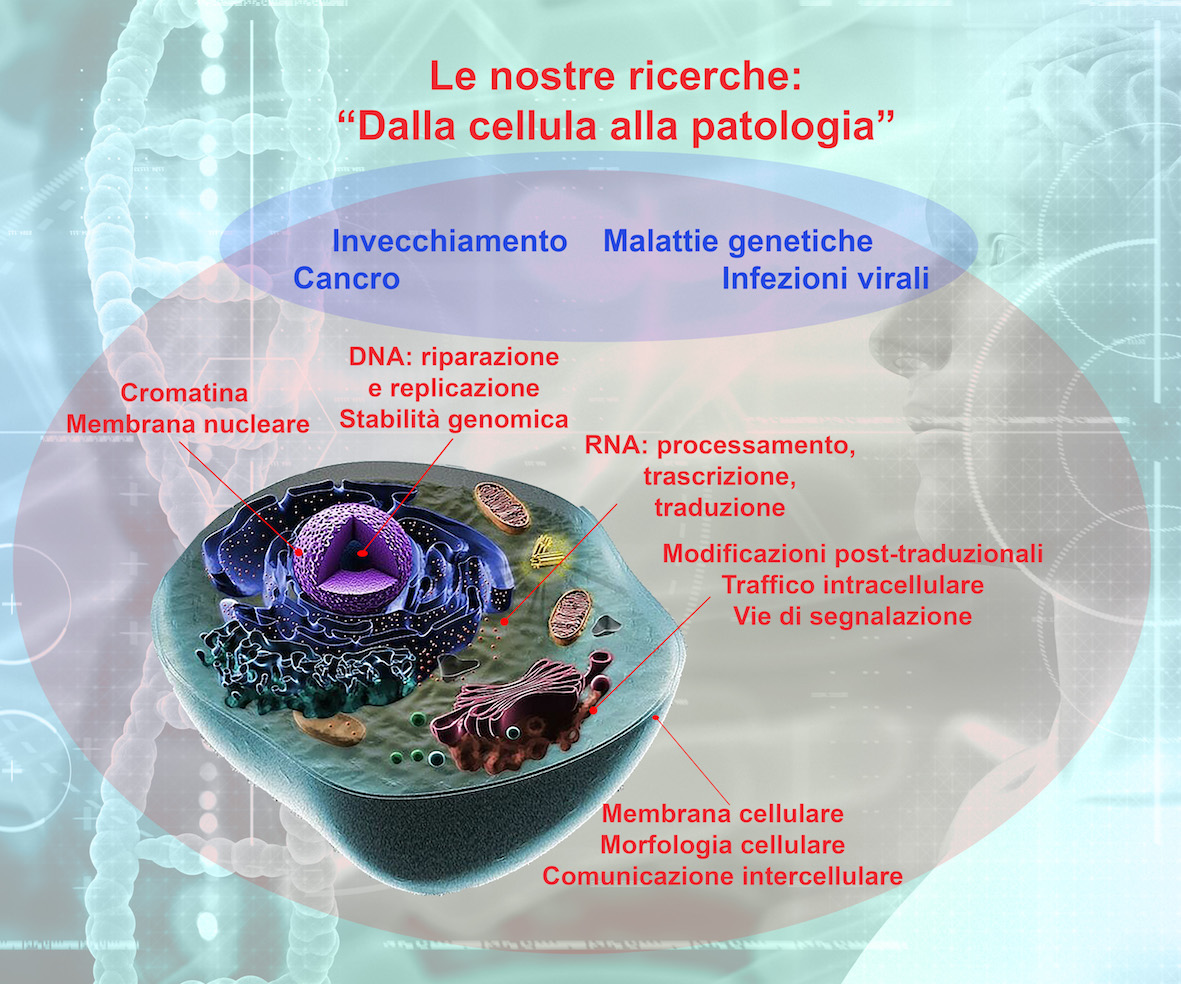
L’Istituto di Genetica Molecolare IGM-CNR “Luigi Luca Cavalli-Sforza” è un centro multidisciplinare per lo studio della cellula sia normale che patologica con un approccio sistemico.
La filosofia che guida la nostra ricerca può essere riassunta come “dalla cellula alla patologia”. Per noi, infatti, lo studio del flusso dell’informazione genetica, dal DNA alle proteine, e delle sue alterazioni patologiche rappresenta la chiave per raggiungere i nostri obiettivi: fornire cure migliori e promuovere la salute e il benessere della società.
La ricerca all’IGM-CNR è focalizzata su alcuni temi centrali:
STABILITA’ DEL GENOMA E TUMORI
Il controllo della qualità e dell’integrità dell’informazione genetica e la sua corretta duplicazione ed espressione sono essenziali per il corretto funzionamento della cellula. All’IGM- CNR si studiano meccanismi di controllo dell’espressione genica (trascrizione, splicing, RNA non codificanti) e della proliferazione (replicazione e riparazione del DNA, ciclo cellulare, differenziamento) di cellule umane sia normali che tumorali, allo scopo di ideare approcci in grado di colpire selettivamente le cellule malate.
Personale coinvolto
Biamonti Giuseppe, Blalock William, Branzei Dana, Buscemi Giacomo, Cenni Vittoria, Chiarini Francesca, Colombi Paolo, Crespan Emmanuele, d’Adda di Fagagna Fabrizio, Evangelisti Camilla, Francia Sofia, Ghigna Claudia, Liberi Giordano, Montecucco Alessandra, Piazzi Manuela, Prosperi Ennio, Sabbioneda Simone, Zadra Giorgia, Zannini Laura
MALATTIE GENETICHE
Mutazioni ereditarie del DNA in grado di alterare l’espressione o la funzione di una proteina sono alla base delle malattie genetiche. I ricercatori dell’IGM-CNR sono impegnati nelle analisi genetiche e funzionali di mutazioni patologiche e identificazione di geni-malattia con particolare enfasi sulle sindromi da difetti della riparazione del DNA e della struttura del nucleo, per trovare nuove strategie per la diagnosi e cura.
L’IGM inoltre coordina il Network Italiano delle Laminopatie.
Personale coinvolto
Andrenacci Davide, Botta Elena, Capanni Cristina, Cenni Vittoria, d’Adda di Fagagna Fabrizio, Evangelisti Camilla, Francia Sofia, Kiriakidu Despina, Lattanzi Giovanna, Mattioli Elisabetta, Nardo Tiziana, Orioli Donata, Peverali Antonio Fiorenzo, Ricotti Roberta, Robuffo Iole, Sabatelli Patrizia, Santi Spartaco, Squarzoni Stefano
VIROCELLULE
Una virocellula è una cellula infetta da un virus, che ne riprogramma il metabolismo per dirigere la sintesi del genoma e delle proteine virali. I ricercatori dell’IGM-CNR sono impegnati nello studio delle complesse interazioni a livello molecolare di diversi virus umani con le cellule ospiti, allo scopo di sviluppare nuovi approcci per combattere le infezioni.
Personale coinvolto
Crespan Emmanuele, Focher Federico, Garbelli Anna, Maga Giovanni
DRUG DISCOVERY E DIAGNOSTICA
L’IGM-CNR ha per missione quella di coniugare la ricerca di base con l’innovazione tecnologica. Per questo i nostri ricercatori sono attivamente impegnati nell’identificazione di bersagli e molecole per la terapia del cancro e delle infezioni virali. Inoltre, all’IGM-CNR si sviluppano nuove tecniche diagnostiche basate su metodiche ottiche/fluorescenti e molecolari. All’IGM sede secondaria di Bologna, si studiano strategie terapeutiche per la progeria di Hutchinson-Gilford, la distrofia muscolare di Emery-Dreyfuss, altre laminopatie e le forme di distrofia muscolare causate da mutazioni del collagene VI (Col6-MD)
Personale coinvolto
Capanni Cristina, Croce Anna Cleta, Crespan Emmanuele, Focher Federico, Garbelli Anna, Grifone Giovanna, Lattanzi Giovanna, Mattioli Elisabetta, Peverali Antonio Fiorenzo, Rapino Monica, Robuffo Iole, Sabatelli Patrizia, Santi Spartaco, Squarzoni Stefano, Zini Nicoletta
ANALISI BIOINFORMATICA DI DATI COMPLESSI
La complessità dei sistemi biologici si riflette in una altrettanto grande complessità dei dati che vengono raccolti sperimentalmente e devono poi essere interpretati. Presso l’IGM-CNR è attivo un gruppo di bioinformatica che sviluppa algoritmi per l’archiviazione, gestione e analisi di grandi moli di dati biologici, come quelli risultanti da analisi genomiche, proteomiche e metabolomiche.
Personale coinvolto
Alfieri Roberta, Biino Ginevra, Bione Silvia, Damiani Giuseppe, Ferrari Francesco, Lisa Antonella